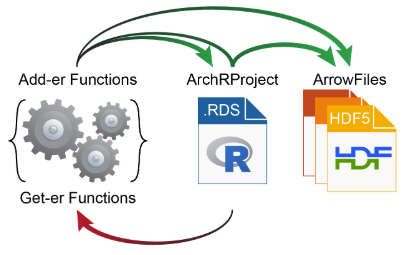
第3章: 创建ArchRProject
ArchRProject对象允许我们将多个Arrow文件整理到单个项目之中。ArchRProject比较小能够直接存放在内存中。通过操作ArchRProject,我们能够快速读取Arrow文件里的数据,并将修改后的数据写回到Arrow文件里。因此你几乎可以在这一章中找到后续分析所要用到的所有函数。此外,由于ArchRProject文件能够进行保存并在之后进行读取,那么也就保证了分析的连续性,同时也方便合作者之间的相互交流。这一章主要介绍如何创建ArchRProject对象并和它进行交互。
3.1 创建一个ArchRProject
ArchRProject至少需要设置两个参数,ArrowFiles和outputDirectory. 其中ArrowFilesArrow文件的文件路径,如果没有,清先阅读第一章将BAM/Fragments文件转成Arrow文件。outputDirectory指的是下游分析得到的文件的保存路径。ArchR会自动将之前提供的geneAnnotation和genomeAnnotation和新的ArchRProject项目进行关联。这些在我们在之前运行addArchRGenome("hg19")的时候被保存在环境变量中。
projHeme1 <- ArchRProject(
ArrowFiles = ArrowFiles,
outputDirectory = "HemeTutorial",
copyArrows = TRUE #This is recommened so that if you modify the Arrow files you have an original copy for later usage.
)
因为这是我们hematopoiesis项目的第一轮分析,所以我们将ArchRProject命名为"proJHeme1"。在后续的分析中,我们会不断修改并更新这个ArchRProject对象,我们需要使用不同的变量名对项目进行跟踪。如果变量名始终都是同一个,那么就可能出现明明是同一个命令,有些时候能运行成功有些时候运行失败的情况,原因可能就是某些中间步骤被漏掉了,导致同一个变量名的背后的实际内容其实是不一样的。
我们可以直接检查下ArchRProject的内容
projHeme1
#输出信息
# class: ArchRProject
# outputDirectory: /path/to/HemeTutorial
# samples(3): scATAC_BMMC_R1 scATAC_CD34_BMMC_R1 scATAC_PBMC_R1
# sampleColData names(1): ArrowFiles
# cellColData names(15): Sample TSSEnrichment ... DoubletEnrichment BlacklistRatio
# numberOfCells(1): 10661
# medianTSS(1): 16.832
# medianFrags(1): 3050
从上面的输入中,我们可以发现ArchRProject在初始化的时候增加了一些额外信息
- outputDirectory: 输出目录
- samples: 样本名
- sampleColData: 记录每个样本的元信息
- cellColData: 记录每个细胞的元信息,第二章计算的"DoubleEnrichment","DoubletScore"就存放在此处
- numberOfCells: 项目总的合格细胞数,也就是不包括之前没有通过质控,或者被认为doublet的细胞
1.medianTSS/medianFrags: 所有细胞的TSS值和Fragments数的中位数
我们可以通过下面这行代码了解下ArchRProject在R中会使用多少内存
paste0("Memory Size = ", round(object.size(projHeme1) / 10^6, 3), " MB")
# "Memory Size = 37.477 MB"
我们还可以检查当前的ArchRProject中存放了哪些矩阵数据,这些数据一旦增加后就可以在下游分析中使用。
getAvailableMatrices(projHeme1)
# "GeneScoreMatrix" "TileMatrix"
3.2 操作ArchRProject对象
既然已经在上一节创建了ArchRProject,这一节就来介绍一些比较常用的操作来从该对象中获取我们需要的信息和保存我们的数据
例1: 使用$访问cellColData
我们可以使用$访问细胞名(cellNames), 样本名(sample)和TSS富集得分(TSS enrichment Scores)
head(projHeme1$cellNames)
head(projHeme1$Sample)
quantile(projHeme1$TSSEnrichment)
除了这些以外,你还可以利用R的补全功能,在输入projHeme1$后看看它还能接哪些内容。
例2: 从ArchRProject中提取部分细胞
我们可以将以前学习的R语言向量/矩阵/数据框提取数据的方法无缝的迁移到ArchRProject上,但区别在于ArchR并不会直接提取数据,而是构建一个新的ArchRProject对象。
最简单的方法是根据位置和细胞名
# 根据位置
projHeme1[1:100, ]
# 根据细胞名
projHeme1[projHeme1$cellNames[1:100], ]
复杂一些就是先根据逻辑判断提取细胞名,接着根据细胞名提取数据。例如挑选scATAC_BMMC_R1的细胞,或是TSSEnrichment大于8的细胞。
# sample name
idxSample <- BiocGenerics::which(projHeme1$Sample %in% "scATAC_BMMC_R1")
cellsSample <- projHeme1$cellNames[idxSample]
projHeme1[cellsSample, ]
# TSS enrichment score
idxPass <- which(projHeme1$TSSEnrichment >= 8)
cellsPass <- projHeme1$cellNames[idxPass]
projHeme1[cellsPass, ]
例3: 在ArchRProject增加数据
我们可以通过在cellColData增加新列的方式来为我们项目中的细胞增加额外的元信息。
例如,我们可以从原来的样本名中提取更易阅读的部分添加到cellColData中。
bioNames <- gsub("_R2|_R1|scATAC_","",projHeme1$Sample)
head(bioNames)
#[1] "BMMC" "BMMC" "BMMC" "BMMC" "BMMC" "BMMC"
一种方式是使用$直接往cellColData添加列
projHeme1$bioNames <- bioNames
或者是用addCellColData()函数往cellColData中添加新的列。使用addCellColData()的好处在于,你可以只为部分增加信息。
bioNames <- bioNames[1:10]
cellNames <- projHeme1$cellNames[1:10]
projHeme1 <- addCellColData(ArchRProj = projHeme1, data = paste0(bioNames),
cells = cellNames, name = "bioNames2")
ArchR默认会用NA填充其余部分。正因如此,如果我们对比bioNames和bioNames2时,你就可以发现NA填充了那些没有被选择的部分
getCellColData(projHeme1, select = c("bioNames", "bioNames2"))
DataFrame with 10661 rows and 2 columns
# bioNames bioNames2
# <character> <character>
#scATAC_BMMC_R1#TTATGTCAGTGATTAG-1 BMMC BMMC
#scATAC_BMMC_R1#AAGATAGTCACCGCGA-1 BMMC BMMC
#scATAC_BMMC_R1#GCATTGAAGATTCCGT-1 BMMC BMMC
#scATAC_BMMC_R1#TATGTTCAGGGTTCCC-1 BMMC BMMC
#scATAC_BMMC_R1#TCCATCGGTCCCGTGA-1 BMMC BMMC
#... ... ...
#scATAC_PBMC_R1#GCTGCGAAGATCCGAG-1 PBMC NA
#scATAC_PBMC_R1#GCAGCTGGTGGCCTTG-1 PBMC NA
#scATAC_PBMC_R1#GCAGATTGTACGCAAG-1 PBMC NA
#scATAC_PBMC_R1#TTCGTTACATTGAACC-1 PBMC NA
#scATAC_PBMC_R1#CGCTATCGTGAGGTCA-1 PBMC N
例4: 从cellColData中提取列
ArchR提供了getCellColData用于从ArchRProject中提取元信息列。你可以认为这是$的功能增强版,因为$只能提取一列,而getCellColData可以提取多列信息,此外还有许多神奇的操作。
例如我们可以根据列名获取数据,比方说每个细胞的唯一fragment数
df <- getCellColData(projHeme1, select = "nFrags")
甚至,你还能在列名中添加一些运算操作,这样会直接输出运算后的结果
df <- getCellColData(projHeme1, select = c("log10(nFrags)", "nFrags - 1"))
例5: 绘制质控信息和TSS富集得分的对比图
根据上述提供的案例,我们可以很容易获取到每个细胞的标准scATAC-seq质控信息。我们发现目前最靠谱的质控标准是TSS富集的得分(评估ATAC-seq数据的信噪比)和唯一比对数(比对数如果不够,那么该细胞也没有分析的价值)
我们先用getCellColData提取我们需要的两列,其中nFrages需要进行log10运算
df <- getCellColData(projHeme1, select = c("log10(nFrags)", "TSSEnrichment"))
接下来,我们基于TSS富集得分和唯一比对进行绘图,函数ggPoint是ArchR对ggplot2的点图相关函数的封装。
p <- ggPoint(
x = df[,1],
y = df[,2],
colorDensity = TRUE,
continuousSet = "sambaNight",
xlabel = "Log10 Unique Fragments",
ylabel = "TSS Enrichment",
xlim = c(log10(500), quantile(df[,1], probs = 0.99)),
ylim = c(0, quantile(df[,2], probs = 0.99))
) + geom_hline(yintercept = 4, lty = "dashed") + geom_vline(xintercept = 3, lty = "dashed")
p

该图的主要作用是找到高质量的细胞。你可能会注意到一些细胞已经在我们之前创建Arrow文件时被设置的阈值filterTSS, filterFargs过滤掉了。然而,如果我们觉得之前的质控过滤对于这个样本不太合适,我们可以根据该图调整阈值,有必要的话还可以重新产生Arrow文件。
尽管可以使用PDF的方式保存上图,但是对于这种点特别多的图,还是用PNG形式比较好。
png("TSS-vs-Frags.png")
plot(p)
dev.off()
getwd()
3.3 为ArchRProject每个样本绘制统计信息
如果一个项目中有多个不同的样本,我们很自然地就会想比较这些样本。所谓一图胜千言,ArchR提供了两种小提琴图(violin plot)和山脊图(ridge plot)用来展示不同组之间的信息。这两种类型的图可以用一个函数plotGroups进行绘制。当然这函数除了根据样本进行分组绘图外,还可以使用下游分析得到的分组信息(例如聚类)。
例1: 根据TSS富集得分为每个样本绘制山脊图。
绘制山脊图时,需要设置plotAs = "ridges"
p1 <- plotGroups(
ArchRProj = projHeme1,
groupBy = "Sample",
colorBy = "cellColData",
name = "TSSEnrichment",
plotAs = "ridges"
)
p1

例2: 根据TSS富集得分为每个样本绘制小提琴图。ArchR绘制的小提琴图额外叠加了一层箱线图(a box-and-whiskers plot in the style of Tukey),中间水平的三条线表示数据中的1/4分位数,中位数和3/4分位数。最下方是最小值,最上方是最大值(或者是1.5倍的IQR, interquartile range, 1/4分位数与3/4分位数的距离)
绘制小提琴图时,需要设置plotAs = "violin"
p2 <- plotGroups(
ArchRProj = projHeme1,
groupBy = "Sample",
colorBy = "cellColData",
name = "TSSEnrichment",
plotAs = "violin",
alpha = 0.4,
addBoxPlot = TRUE
)

例3: 根据log10(unique nuclear fragments)为每个样本绘制山脊图。
p3 <- plotGroups(
ArchRProj = projHeme1,
groupBy = "Sample",
colorBy = "cellColData",
name = "log10(nFrags)",
plotAs = "ridges"
)
p3

例4: 根据log10(unique nuclear fragments)为每个样本绘制小提琴图。
p4 <- plotGroups(
ArchRProj = projHeme1,
groupBy = "Sample",
colorBy = "cellColData",
name = "log10(nFrags)",
plotAs = "violin",
alpha = 0.4,
addBoxPlot = TRUE
)
p4

我们可以使用plotPDF()将上面的图绘制在一个PDF中
plotPDF(p1,p2,p3,p4, name = "QC-Sample-Statistics.pdf", ArchRProj = projHeme1, addDOC = FALSE, width = 4, height = 4)
3.4 绘制样本的TSS富集谱和Fragment大小分布
鉴于数据的存储和读取方式, ArchR能够快速的从Arrow文件中计算出fragment大小分布和TSS富集谱。
Fragments大小分布: 我们使用plotFragmentSizes函数为绘制所有样本的fragment大小分布。ATAC-seq数据中的fragment大小分布可能会因样本、细胞类型和批次不同,但这和数据质量并不是严格相关。比如说我们的数据在不同组之间就存在一些差别。
p1 <- plotFragmentSizes(ArchRProj = projHeme1)
p1

TSS富集谱: 我们使用plotTSSEnrichment()函数绘制TSS富集谱。TSS富集谱需要在中心位置有一个明显的峰,在峰的右边还会有一个核小体引起的小隆起(约147 bp)。
p2 <- plotTSSEnrichment(ArchRProj = projHeme1)
p2

和之前一样,我们可以使用plotPDF()将图形以可编辑的矢量图进行保存。
plotPDF(p1,p2, name = "QC-Sample-FragSizes-TSSProfile.pdf", ArchRProj = projHeme1, addDOC = FALSE, width = 5, height = 5)
3.5 保存和加载ArchRProject
ArchR提供了saveArchRProject函数让我们能将内存中ArchRProject对象的保存到本地,并在之后进行读取,当然保存到本地的数据也就能分享给其他人。从根本上来讲,ArchRProject是一个指向多个Arrow文件的对象,因此使用saveArchRProject()保存ArchRProject的过程实际上分为两步
- 将当前的Arrow文件复制到目标输出目录下
outputDirectory,确保它们能和新的ArchRProject关联 - 将目标
ArchRProject保存到outputDirectory目录下
我们这里以projHeme1为例介绍saveArchRProject()的用法
saveArchRProject(ArchRProj = projHeme1, outputDirectory = "Save-ProjHeme1", load = FALSE)
运行结束后,outputDirectory对应的目录下就会保存相应的Arrow文件和projHeme1的Rds文件。
重要知识点: 这一步骤不会自动更新你当前R session里活跃的ArchRProject对象。也就说,当前R session里的projHeme1对象还是指向原来的Arrow文件,而不会指向拷贝到outputDirectory目录下的Arrow文件。
换言之,如果我们想要将当前的项目复制到新的目录,你可以设置load=TRUE。下面的代码运行结束后得到的projHemeNew就是指向拷贝后Arrow文件的ArchRProject对象。你可以使用原来的projHeme1,这就会覆盖当前R session的projHeme1。
projHemeNew <- saveArchRProject(ArchRProj = projHeme1, outputDirectory = "Save-ProjHeme1", load = TRUE)
3.6 使用ArchRProject过滤doublets
当我们使用addDOubletScores()增加了预测的doublet信息后,我们就能使用filterDoublets()过滤这些预测的doublets。这个函数里的一个关键参数是filterRatio, 由于不同样本的制备过程中的细胞上样量不同,那么不同样本的doublet也就不同,为了保证不同样本的过滤后结果保持一致,所以引入了filterRatio。该值越高,被认为是doublet而被过滤的细胞也就越多。
了解了filterRatio的作用后,我们再来看它的定义,它是根据未被过滤的细胞数计算的需要被过滤doublet的最大值(the maximum ratio of predicted doublets to filter based on the number of pass-filter cells)。举个例子。假如这里有5000个细胞,需要被过滤doublet的最大值就等于 filterRatio * 5000^2 / (100000), 也就是filterRatio * 5000 * 0.05。
如果还是不太了解,也不太纠结,毕竟大部分时候我们用的都是默认参数。我们现在只需要知道这个filterRatio参数很重要,得根据后续结果进行调整。这里就直接运行filterDoublets对细胞进行过滤。出于教学目的,我们将处理结果保存为一个新的ArchRProject对象,也就是projHeme2,实际分析中,你可以使用原来的名字覆盖之前的结果,降低内存消耗。
projHeme2 <- filterDoublets(projHeme1)
# Filtering 410 cells from ArchRProject!
# scATAC_BMMC_R1 : 243 of 4932 (4.9%)
# scATAC_CD34_BMMC_R1 : 107 of 3275 (3.3%)
# scATAC_PBMC_R1 : 60 of 2454 (2.4%)
之前的projHeme1有10,661个细胞,经过上一步的过滤,projHeme2剩下了10,251个细胞,也就意味着有410个细胞(3.85%)是doublet。而且每个样本的过滤比例并不相同,这就是filterRatio的作用。
projHeme2
# ...
# numberOfCells(1): 10251
# medianTSS(1): 16.856
# medianFrags(1): 2991
如果你想过滤更多ArchRProject里的细胞,你可以设置更高的filterRatio。和参数调整的更多信息可以用?filterDoublets查看。
projHemeTmp <- filterDoublets(projHeme1, filterRatio = 1.5)
# Filtering 614 cells from ArchRProject!
# scATAC_BMMC_R1 : 364 of 4932 (7.4%)
# scATAC_CD34_BMMC_R1 : 160 of 3275 (4.9%)
# scATAC_PBMC_R1 : 90 of 2454 (3.7%)
最后,既然projHemeTmp只是说明用的临时对象,我们现在可以将其从R session中删除了。
rm(projHemeTmp)
