第5章: 使用ArchR聚类
大部分单细胞聚类算法都在降维后空间中计算最近邻图,然后鉴定"社区"或者细胞聚类。这些方法效果表现都特别出色,已经是scRNA-seq的标准策略,所以ArchR直接使用了目前scRNA-seq包中最佳的聚类算法用来对scATAC-seq数据进行聚类。
5.1 使用Seurat的FindClusters()函数进行聚类
我们发现Seurat实现的图聚类方法表现最好,所以在ArchR中,addClusters()函数本质是上将额外的参数通过...传递给Seurat::FindClusters()函数,从而得到聚类结果。在分析中,我们发现Seurat::FindClusters()是一个确定性的聚类算法,也就是相同的输入总是能得到完全相同的输出。
projHeme2 <- addClusters(
input = projHeme2,
reducedDims = "IterativeLSI",
method = "Seurat",
name = "Clusters",
resolution = 0.8
)
# 只展示部分信息
# Maximum modularity in 10 random starts: 0.8568
# Number of communities: 11
# Elapsed time: 1 seconds
我们可以使用$符号来获取聚类信息,输出结果是每个细胞对应的cluster
head(projHeme2$Clusters)
# [1] "C10" "C6" "C1" "C2" "C2" "C10"
我们统计下每个cluster的细胞数
table(projHeme2$Clusters)
# C1 C10 C11 C2 C3 C4 C5 C6 C7 C8 C9
# 310 1247 1436 480 323 379 852 1271 677 2550 726
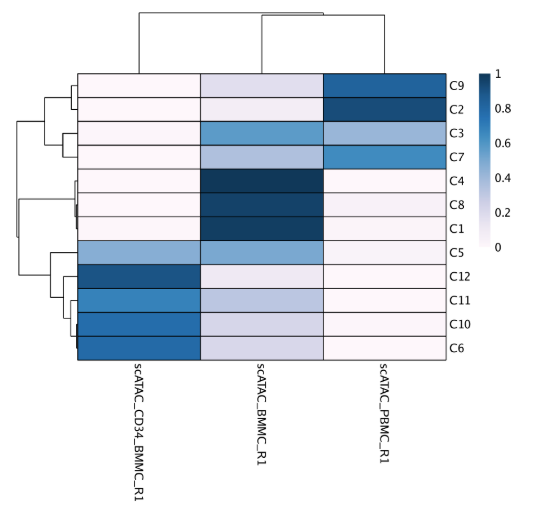
为了更好了解样本在cluster的分布,我们可以使用confusionMatrix()函数通过每个样本创建一个聚类混淆矩阵(cluster confusion matrix)
cM <- confusionMatrix(paste0(projHeme2$Clusters), paste0(projHeme2$Sample))
cM
文字信息太多,这里直接用热图进行展示
library(pheatmap)
cM <- cM / Matrix::rowSums(cM)
p <- pheatmap::pheatmap(
mat = as.matrix(cM),
color = paletteContinuous("whiteBlue"),
border_color = "black"
)
p

细胞有时在二维嵌入中的相对位置与所识别的聚类并不完全一致。更确切的说,单个聚类中的细胞可能出现在嵌入的多个不同区域中。在这些情况下,可以适当地调整聚类参数或嵌入参数,直到两者之间达成一致。
5.2 使用scran聚类
除了Seurat, ArchR还能够使用scran进行聚类分析,我们只需要修改addClusters()中的method参数即可。
projHeme2 <- addClusters(
input = projHeme2,
reducedDims = "IterativeLSI",
method = "scran",
name = "ScranClusters",
k = 15
)
