唐海宝老师开发的JCVI有一个工具,叫做ALLMAPS, 能够展示遗传图谱和物理图谱的对应关系,如下所示
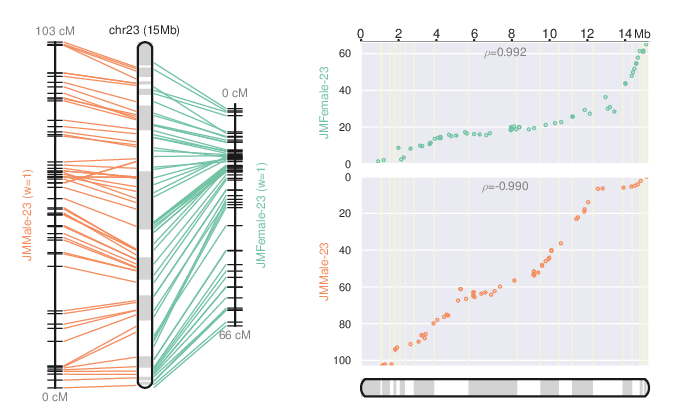
但是这个图的目标是为了对ALLMAPS的scaffold结果进行可视化,并不是专门用于展示遗传图谱的标记和物理图谱的对应关系。尽管在allmaps这个组件下提供了plot函数,命令行输入只要求 input.bed 和 seqid, 但实际运行的时候还要求 allmaps path的中间文件, xxxx.lifted.bed, xxxx.agp, weight.txt等文件。
为了解决这一问题,我阅读了allmaps.py的源代码,在plot的基础上增加了一个plot2函数,只需要用户输入 input.bed, 染色体编号和染色体的长度就能够画图。
# python -m jcvi.assembly.allmaps plot2 input.bed 染色体编号 染色体长度
python -m jcvi.assembly.allmaps plot2 input.bed chr1 123140023
其中input.bed的格式要求有6列,
- 标记所在染色体名
- 标记所在染色体的start
- 标记所在染色体的end, 通常就是start+1
- 标记对应的图谱位置, 要求输入为"图谱名-连锁图谱所在组:连锁图谱的遗传距离"
- 标记名
案例
Chr1 68185909 68185910 male-14:48.470000 Chr1:68185910
Chr1 68479621 68479622 male-14:49.380000 Chr1:68479622
Chr1 68595299 68595300 male-14:48.440000 Chr1:68595300
可以先生成如下的csv文件,然后转换成bed
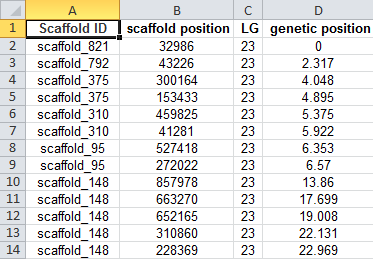
python -m jcvi.assembly.allmaps merge male.csv male.bed
目前有plot2函数的代码还在我的项目下,xuzhougeng/jcvi, 待代码稳定了,再PR。
