在我大学的时候,构建遗传图谱靠的是人工跑胶,然后看胶图统计基因型。当时我用的SSCP(单分子构象多态性)技术区分单个碱基存在差异的等位基因,要放在4度过夜12小时,然后第二天银染显色,放在一个医学看片的设备上读条带。
现在测序便宜了,简化基因组测序随随便便就能获得成千上万的分子标记。然而标记多有标记多的烦恼,就是以前的作图软件不好用了。以前的暴力穷举的方法在海量标记面前,几乎不可能在有限的时间里完成构图任务,而且更加麻烦的是高通量的SNP标记还容易出错。
R语言中有一些建图软件包,不过都有一些问题。qtl包的建图算法太老,onemap的算法是SERIATION, RECORD, RCD, UG, 但是代码不是C/C++写的,所以运行速度太让人着急。
在2017年,出现了一个新的R包,ASMap,它其实就是将MSTmap移植到了R语言中。MSTmap是很久之前就提出的高通量作图方法,只不过需要编译,有点不太友好。
ASMap支持如下群体:
- BC: 回交群体
- DH: 双单倍体群体
- ARIL: 高世代重组近交系, 理论上是已经纯合的遗传群体(除了部分剩余杂合位点), 属于永久性群体
- RILn: 低世代重组近交系,n=2时,就是F2群体。
算法说明简分为两个部分:
-
聚类算法: 如果分子标记 $m_j$ 和$m_k$ 来自于两个不同的连锁群,那么$P_=0.5$, 并且标记间的加权距离(hamming distance)为 $E(d_) = n/2$。 根据定义,MSTmap使用Hoeffding不等式确定标记是否来自于同一个连锁群
-
标记排序: 先根据遗传距离对标记进行分箱,也就是将共分离的标记当做一个对象。之后排序问题就视作 TSP(旅行商问题), 也就是寻找所有标记间最短的路径。
可用函数
图谱构建函数: 一共有两个,注意p.value的调整
- mstmap.data.frame()
- mstmap.cross(): 从qtl包的对象进行转换,对于高世代的RIL群体,要用
conver2riself转换类型, 对于BCnFn群体要用conver2bcsft转换类型
标记调整函数:
- pullCross(): 抽取异常分子标记
- pushCross(): 检查完毕后,将符合要求的标记放回原处
- pp.init(): 调整偏分离的阈值
可视化诊断:
- profileGen(): 统计基因型中交换数,双交换数,缺失情况。 适用于对遗传图谱质量进行评估
- profileMark: 统计标记的偏分离,缺失比例,等位基因分布,双交换数
- heatMap(): 以标记间RF(重组率)绘制热图
使用案例
内容翻译自: R Package ASMap: Effcient Genetic Linkage Map Construction and Diagnosis
第一步: 环境准备
加载环境和数据,其中数据集为300个群体,3023个标记的回交群体
library("qtl")
library("ASMap")
data("mapBCu")
summary(mapBCu)
如果是自己的数据集,有两种方法可以构建,一种是用mstmap.cross 转换qtl包中read.cross得到的对象,如下
mapthis <- read.cross("csv", "http://www.rqtl.org/tutorials", "mapthis.csv",
estimate.map=FALSE)
mapthis <- convert2bcsft(mapthis, BC.gen = 0, F.gen = 2, estimate.map = FALSE)
mapthis <- mstmap.cross(mapthis, id = "id")
或者用mstmap.data.frame, 要调整如下的参数:
- object: 数据框, 行对应的标记名, 列是每个个体对应的基因型. 注, 构建数据框时,一定要保证字符串不能被当做是因子(stringsAsFactors=FALSE)。
- pop.type: 群体类型,“BH","BC", "RILn"(低世代的近交系,RIL2可以认为是F2), "ARIL"(高世代的近交系)
- dist.fun: 计算遗传距离的函数, 默认是"kosambi", 不需要更改。
- p.value: 和群体大小有关,群体越大,p值要越小,也就是要更严格。
对于低世代的近交系,亲本基因型编码为"A"或"a", "B"或"b", 杂合基因型编码为"X". 所谓的缺失值编码为"U"或"-"
mstmap.data.frame这一步运行会比较慢,这是由于它会预先对数据进行分箱,减少标记量,提高后续的运算效率。

第二步: 预处理
在正式构建遗传图谱之间,我们需要谨慎的按照如下清单检查我们用于构图的基因型/分子标记的质量,包括但不限于
- 检查每个个体对应的标记缺失比例,以及每个标记在所有个体中的缺失失比例。缺失比例过高说明标记或个体存在问题
- 检查群体间是否存在标记高度相似的两个个体。
- 检查过度偏分离(segregation distortion)的标记。 高度偏分离的标记可能不会定位到唯一的座位上。
- 检查交换等位基因的标记。这些标记在构建图谱期间,难以聚类,或者和其他标记有关联,因此需要在分析前修复他们的匹配。
- 检查共分离的标记。 共分离的标记会严重影响构图时的计算效率,所以可以先暂时把它们忽略掉。
检查每个个体的基因型缺失情况
plot.missing(mapBcu)

水平的黑线表示一些个体中,大部分的标记都没有对应的基因型,因此我们将这些个体移除。
sg <- statGen(mapBCu, bychr=FALSE, stat.type = "miss")
mapBC1 <- subset(mapBCu, ind=sg$miss < 1600)
从图谱构建的角度来看,高度相关的个体会增强标记的偏分离. 因此对于基因型几乎一模一样的个体,最好在建立图谱前移除
gc <- genClones(mapBC1, tol=0.95)
gc$cgd
从R返回的结果中,可以发现有将近11组的基因型几乎一样。但是第一组的BC045和BC039由于存在大量的基因型缺失,不足以说明两者相同,同样情况的还有BC052和BC045,以及BC168和BC045, 可以排除这三项。
cgd <- gc$cgd[-c(1,4,5)]
mapBC2 <- fixClones(mapBC1, cgd, consensus = TRUE)
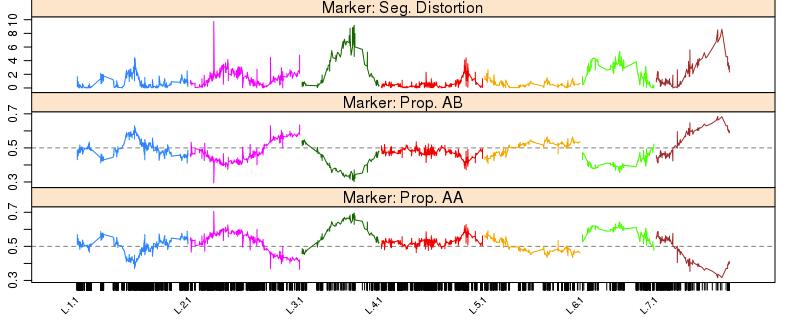
接下来,可以进一步检查特定位点的观测等位基因频率和期望频率的偏差。这有可能是基因型考察错误,也有可能意味着该区域受到潜在的生物学和遗传学机制影响。这一步,同时检查分子标记的偏分离情况,不同基因型的占比,和缺失率。
profileMark(mapBC2, stat.type = c("seg.dist", "prop","miss"),
crit.val = "bonf", layout = c(1,4), type="l")

对于高度的偏分离的标记,我们应该直接忽略掉
mm <- statMark(mapBC2, stat.type = "marker")$marker$AB
dm <- markernames(mapBC2)[(mm > 0.98) | (mm < 0.2)]
mapBC3 <- drop.markers(mapBC2, dm)
而对于不偏分离不怎么严重的标记,最好的方法是先把他们放在一边,等图谱构建完成之后再把它们放回到遗传图谱中。
pp <- pp.init(miss.thresh = 0.1, seg.thresh = "bonf")
mapBC3 <- pullCross(mapBC3, type = "missing", pars=pp)
mapBC3 <- pullCross(mapBC3, type="seg.distortion", pars=pp)
mapBC3 <- pullCross(mapBC3, type="co.located")
第三步: 构图
构图通常不是一次性完成的工作,需要多次迭代。先进行第一次尝试,运行时间取决于计算机性能和标记数目
mapBC4 <- mstmap(mapBC3, bychr = FALSE, trace = TRUE, p.value = 1e-12)
chrlen(mapBC4) #每个连锁群的长度
对构建出图谱进行可视化检查
heatMap(mapBC4, lmax = 70)

热图分为两个部分看。上三角展示的是标记间的重组率,下三角则是标记间的LOD值,两者模式要是一样,那就说明图谱还是靠谱的。中间的正方形为连锁不平衡区(LD block), 一般在连锁不平衡区极少发生交换,而不同的连锁不平衡区交换频繁。
但是光看热图还是不够的,我们还得保证遗传图谱里个体的交换次数不应该太多。也就是,每个个体能发生的交换是有限的,比如说如果染色体长度为200cM,那么在回交后代中,每个个体的重组次数应该不多于14次。
水稻每条染色体平均0.5~1次交换。人类每次减数分裂平均30次。
pg <- profileGen(mapBC4, bychr = FALSE, stat.type = c("xo", "dxo", "miss"),
id="Genotype", xo.lambda = 14, layout=c(1,3),lty=2)

从上图可以发现,一些个体的重组次数明显过高,而且这些个体的双交换次数明显多于其他个体,缺失率也是如此。
我们需要将这些个体剔除,对遗传图谱进行升级
mapBC5 <- subsetCross(mapBC4, ind = !pg$xo.lambda)
mapBC6 <- mstmap(mapBC5, bychr = TRUE, trace = TRUE, p.value = 1e-12)
chrlen(mapBC6)
和上次相比,每个连锁群的长度明显减少,一般而言遗传图谱越小,图谱越可靠。
profileMark(mapBC6, stat.type = c("seg.dist", "prop", "dxo", "recomb"),
layout = c(1, 5), type = "l")

最后结果表明,每个个体的双交换次数都没有超过1 个。
第四步: 加入前提剔除的标记
对于前期放在一边的标记,可以在图谱构建完成后放回到完成的图谱中,同时要仔细的检查。
先将515个缺失率在10%~20%的标记塞到图谱上。这一步是将当前的标记和图谱上的标记计算重组率,选择最近位置插入。然后展示长度明显小于其他连锁群的4个连锁群,判断是否能够将他们进行合并。
pp <- pp.init(miss.thresh = 0.22, max.rf = 0.3)
mapBC6 <- pushCross(mapBC6, type = "missing", pars = pp)
heatMap(mapBC6, chr = c("L.3", "L.5", "L.8", "L.9"), lmax = 70)

从图中,我们可以发现,L.3和L.5之间有明显的连锁,L8和L.9有明显的连锁。我们使用mergeCross()将这两个连锁群进行合并,这个时候的mstmap要保证bychr=TRUE, 对不同的连锁群分别计算标记距离,而不是从聚类开始。
mlist <- list("L.3" = c("L.3", "L.5"), "L.8" = c("L.8", "L.9"))
mapBC6 <- mergeCross(mapBC6, merge = mlist)
names(mapBC6$geno) <- paste("L.", 1:7, sep = "")
mapBC7 <- mstmap(mapBC6, bychr = TRUE, trace = TRUE, p.value = 2)
chrlen(mapBC7)
这样子就得到了更加优化的图谱。但是L1, L2, L.4的连锁群距离有一些轻微的提高,说明可能存在过多的交换现象
pg1 <- profileGen(mapBC7, bychr = FALSE, stat.type = c("xo", "dxo","miss"),
id = "Genotype", xo.lambda = 14, layout = c(1,3), lty = 2)

我们发现新引入的部分个体是导致该现象的元凶,这就需要我们将这些株系次移除掉,然后重建图谱
mapBC8 <- subsetCross(mapBC7, ind=!pg1$xo.lambda)
mapBC9 <- mstmap(mapBC8, bychr = TRUE, trace = TRUE, p.value = 2)
chrlen(mapBC9)
移除的株系不会对连锁群的数目造成影响,连锁图谱只会在组内进行重建。大部分连锁群的长度都有了一定程度的下降。继续观察一下标记的遗传图谱的表现情况
profileMark(mapBC9, stat.type = c("seg.dist", "prop"), layout = c(1, 5),
type = "l")

从图中可以发现,在L.2里面有一个诡异的突起(偏分离), 需要被移除。当然我们也能看到在L.3, L.5, L.7中,新增的缺失率10%~20%的标记,有一定程度的偏分离。剩下的295个标记为"seg.distortion"的标记说不定能解释这个情况,所以下一步就是把它们塞回去
sm <- statMark(mapBC9, chr = "L.2", stat.type = "marker")
dm <- markernames(mapBC9, "L.2")[sm$marker$neglog10P > 6]
mapBC10 <- drop.markers(mapBC9, dm)
pp <- pp.init(seg.ratio = "70:30")
mapBC11 <- pushCross(mapBC10, type = "seg.distortion", pars = pp)
mapBC12 <- mstmap(mapBC11, bychr = TRUE, trace = TRUE, p.value = 2)
round(chrlen(mapBC12) - chrlen(mapBC9), 5)
nmar(mapBC12) - nmar(mapBC10)
最后一步就是加入共分离的标记
mapBC <- pushCross(mapBC12, type = "co.located")
最后结果如下

