第1章: ArchR基础入门
这一章将会介绍如何导入数据,如何构建Arrow文件,这是后续ArchR分析的基础。
1.1 ATAC-seq术语介绍
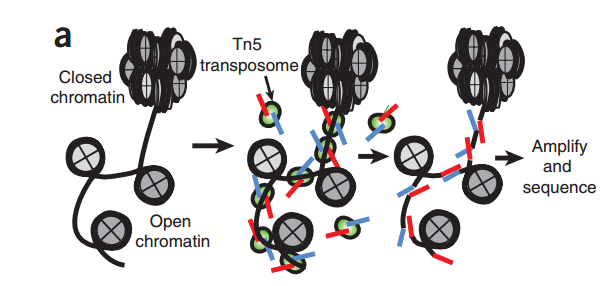
"fragment"是ATAC-seq实验中的一个重要概念,它指的是通过Tn5转座酶对DNA分子进行酶切,然后经由双端测序得到的序列。

我们会根据Tn5插入导致的偏移从read比对得到的位置推断出fragment的起始和结束位置。根据之前的报道,Tn5转座酶以同源二聚体的形式结合到DNA上,在两个Tn5分子间隔着9-bp的DNA序列。根据这个情况,每个Tn5同源二聚体的结合事件会产生2个Insertions,中间隔着9bp。因此,真实的"开放"位置的中心在Tn5二聚体的正中间,而不是Tn5的插入位置。为了尽可能的还原真实情况,我们对Tn5的Insertions进行了校正,即正链的插入结果往右移动4bp(+4 bp), 负链的插入结果往左偏移5bp(-5 bp)。这个和最早提出的ATAC-seq里的描述是一致的。因此,在ArchR中,"fragment"指的是一个table或genomic ranges对象, 记录在染色体上,经过偏移校正后的单碱基起始位置,以及经过偏移校正后单碱基结束位置,每个fragment都会对应唯一的细胞条形码。类似的,"Insertions"这得是偏移校正后的单碱基位置,它位于开放位置的正中心。
"fragment"和"insertions"这两个词我并没有将其翻译成中文,我觉得这两个单词可能就和PCR一样,当说到它们的时候,脑中会有一个画面描述它们,而不是局限一个词。
1.2 为什么是用ArchR
现在其实已经有一些工具能够处理单细胞ATAC-seq数据,我们为什么要额外造一个轮子,开发一个新的项目呢?主要是ArchR提供了其他工具目前尚不能实现的功能

不仅如此,ArchR通过优化数据结构降低了内存消耗,使用并行提高了运行速度,因此保证其性能优于其他同类型工具。在超过70,000个细胞的分析项目中,一些软件需要超过128G的可用内存。

ArchR最初就是根据Unix的笔记本进行设计(我觉得他说的是MacBook Pro),因此对于中等大小的实验(小于100,000个细胞),ArchR能够保证一些特殊分析的运行速度,并能实时的展示结果,让我们能够更深入的和数据进行互动,给出有意义的生物学解释。当然,如果细胞数更多,你最好使用服务器进行分析。ArchR提供了方便的图形导出功能,在服务器处理完项目之后,可以直接下载到本地进行使用。
目前,ArchR并没有针对Windows进行优化。这句话的意思是指,ArchR的并行策略是基于Unix系统而非Windows系统,因此上述说的性能提升不包括Windows。
1.3 什么是Arrow文件/ArchRProject
正如开头所说,ArchR分析项目的基础是Arrow文件。每个Arrow文件记录着每个独立样本的所有相关信息(例如元信息、开放的fragment和数据矩阵)。这里说的独立样本最好是最详尽的分析单元(例如,一种特定条件下的单个重复)。在创建Arrow文件以及一些附加分析中,ArchR会编辑和更新相应的Arrow文件,在其中添加额外的信息层。值得注意的是,Arrow文件实际指的是磁盘上的文件路径。更确切的说,Arrow文件并不是一个存放在内存中的R语言对象,而是存放在磁盘上HDF5文件。正因如此,我们使用ArchRProject对象用来关联这些Arrow文件,将其关联到单个分析框架中,从而保证在R中能高效访问它们。而这个ArchRProject对象占用内存不多,因此才是存放在内存中的R语言对象。

有一些操作会直接修改Arrow文件,而一些操作会先作用于ArchRproject,接着反过来更新每个相关Arrow文件。因为Arrow文件是以非常大的HDF5格式存放,所以ArchR的get-er函数通过和ArchRProject进行交互获取数据,而add-er函数既能直接在Arrow文件中添加数据,也能直接在ArchRpoject里添加数据,或者通过ArchRpoject向Arrow文件添加数据。

1.4 ArchR的输入文件类型
ArchR主要以scATAC-seq原始数据经上游处理后的两种常见输出文件(BAM, fragment)作为输入。Fragment文件记录着scATAC-seq的fragment以及对应的细胞ID,每一行都是一条记录,该文件需要是tabix(见注1)排序并建立索引保证能被高效读取。BAM文件则是二进制格式下的tabix排序文件,记录着scATAC-seq的fragment、原始数据、细胞条形码和其他信息。具体使用何种文件作为输入取决于你的上游处理流程。以10XGenomics的CellRanger软件为例,它的输出文件是fragments,而sci-ATAC-seq流程则输出BAM文件。
ArchR使用scanTabix函数读取fragment文件,使用scanBAM读取BAM文件。输入过程并不会直接读取所有数据,而是每次读取一大块(chunks),然后将这一块数据转换成fragment格式(见注2),经过压缩先暂时以HDF5格式保存到本地磁盘上,避免消耗过多的内存。最后,等所有数据都加载完成之后,该数据相关的所有临时HDF5文件会被重新读取,经过组织之后以单个HDF5形式保存为Arrow文件。ArchR之所以能以较小的内存量高效地读取大文件,就是因为它采用的是这种分块处理的方法,由于每一块数据的处理都互不干扰,因此也就能够并行计算。
- 注1: 当以制表符记录基因组位置信息时,我们可以通过压缩让其体积变小(bgzip/gzip),通过建立tabix索引高效访问给定位置的信息。
- 注2: fragments一共有5列,分别是chromosome, offset-adjusted chromosome start position, offset-adjusted chromosome end position, and cellular barcode ID, duplicate count
1.5 开始设置
在后续分析之前,请先设置好我们的工作目录,设置将要使用的线程数,加载我们的基因和基因组注释。由于每个人的环境都不太一样,所以你后续可能需要用addArchRThreads()修改线程数。默认情况下,ArchR使用系统一半的可用线程,你可以手动进行调整。如果你用的是Windows,那么默认都是1,这是因为ArchR的多线程是基于Unix操作系统。
第零步,安装ArchR。目前ArchR托管在GitHub上,因此无法直接用CRAN或者Bioconductor中直接下载安装。
方法1:适用于网络状态好的情况
if (!requireNamespace("devtools", quietly = TRUE)) install.packages("devtools")
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
devtools::install_github("GreenleafLab/ArchR", ref="master", repos = BiocManager::repositories())
方法2: 适用于上述方法多次失败的情况
先从Github上下载项目到本地, 大约有262Mb
git clone https://github.com/GreenleafLab/ArchR.git
然后在 R里面进行安装
BiocManager::install(c("nabor","motifmatchr","chromVAR","ComplexHeatmap"))
install.packages("./ArchR", repo=NULL)
接着安装一些额外包
ArchR::installExtraPackages()
如果安装失败,就手动安装 Seurat, immunogenomics/harmony, immunogenomics/presto, Cairo,shiny, shinythemes, rhandsontable
第一步,我们加载ArchR包。
library(ArchR)
第二步, 我们需要设置ArchR函数的默认线程数。你需要在每个新的R session中都设置该参数,线程多多益善,但是不要超过总线程的3/4。因为线程会和内存挂钩,所以线程数越多,内存相应使用的也越多。
addArchRThreads(threads = 16)
第三步: 我们设置需要使用基因和基因组注释。同样,这也是每个新的R session需要设置的参数。当然,我们需要使用和序列比对阶段相同的基因组版本。对于本教程使用的数据,我们会使用hg19参考基因组。ArchR支持多种基因组注释,并且允许自定义基因组注释。
addArchRGenome("hg19")
基因和基因组注释信息是后续计算TSS富集得分,核小体含量和基因活跃度得分所必需的。同样,你得保证这里选择的基因组版本得和你上游数据处理时用到的基因组版本一致。我们这里设置"hg19"就是因为上数据处理用的是hg19作为参考基因组。除了hg19外,ArchR还提供了"hg38", "mm9", "mm10", 并且允许用户使用createGeneAnnotation和createGenomeAnnotation函数创建其他物种注释。
我们使用addArchRGenome函数无缝地为ArchR提供这些信息。该函数告诉ArchR,在之后的所有分析中,它应该使用定义在ArchRgenome的相关genomeAnnotation和geneAnnotation。每一个原生支持的基因组都包括四个对象(object)
- BSgenome: 记录 染色体坐标信息和染色体序列信息
- GRanges: 记录blacklist, 即对分析没有用设置可能产生干扰的区域
- TxDb: 定义所有基因的位置信息
- OrgDb: 提供基因编号,以及不同基因编号之间的转换
如下是ArchR自带的物种注释的数据来源
ArchR的预编译的hg19基因组用的是BSgenome.Hsapiens.UCSC.hg19, TxDb.Hsapiens.UCSC.hg19.knownGene, org.Hs.eg.db。而黑名单(记录着一直打开的区域,对后续分析没有帮助的位置)则来源于 hg19 v2 blacklist regions 和mitochondrial regions that show high mappability to the hg19 nuclear genome from Caleb Lareau and Jason Buenrostro,用ArchR::mergeGR()函数进行合并
ArchR的预编译的hg38基因组用的是BSgenome.Hsapiens.UCSC.hg38, TxDb.Hsapiens.UCSC.hg38.knownGene, org.Hs.eg.db。而黑名单(记录着一直打开的区域,对后续分析没有帮助的位置)则来源于 hg19 v2 blacklist regions 和mitochondrial regions that show high mappability to the hg38 nuclear genome from Caleb Lareau and Jason Buenrostro,用ArchR::mergeGR()函数进行合并
ArchR的预编译的mm9基因组用的是BSgenome.Mmusculus.UCSC.mm9, TxDb.Mmusculus.UCSC.mm9.knownGene, org.Mm.eg.db。而黑名单(记录着一直打开的区域,对后续分析没有帮助的位置)则来源于 mm9 v1 blacklist regions 和mitochondrial regions that show high mappability to the mm9 nuclear genome from Caleb Lareau and Jason Buenrostro,用ArchR::mergeGR()函数进行合并
ArchR的预编译的mm10基因组用的是BSgenome.Mmusculus.UCSC.mm10, TxDb.Mmusculus.UCSC.mm10.knownGene, org.Mm.eg.db。而黑名单(记录着一直打开的区域,对后续分析没有帮助的位置)则来源于 mm10 v2 blacklist regions 和mitochondrial regions that show high mappability to the mm10 nuclear genome from Caleb Lareau and Jason Buenrostro,用ArchR::mergeGR()函数进行合并
1.5.1: 自定义ArchRGenome
如上所述,一个ArchRGenome需要包括基因组注释和基因注释
为了构建自定义的基因组注释,我们会使用createGenomeAnnotation. 他需要如下2个信息
- BSgenome: 包括基因组的序列信息,你可以通过谷歌查找指定物种的Bioconductor包
- GRanges: 记录基因组中的黑名单区域,用来过滤下游分析中不需要的区间。这不是必须的,毕竟不是所有物种都能有这个文件。 publication on the ENCODE blacklists提供黑名单列表的制作方法。
例如,我们如果需要为Drosophila melanogaster创建一个基因组注释,那么我们需要先下载对应的BSgenome
if (!requireNamespace("BSgenome.Dmelanogaster.UCSC.dm6", quietly = TRUE)){
BiocManager::install("BSgenome.Dmelanogaster.UCSC.dm6")
}
library(BSgenome.Dmelanogaster.UCSC.dm6)
接着,我们从BSgenome对象中创建基因组注释
genomeAnnotation <- createGenomeAnnotation(genome = BSgenome.Dmelanogaster.UCSC.dm6)
查看这个对象,你可以看到一个ArchR基因组注释的组成内容
genomeAnnotation
## List of length 3
## names(3): genome chromSizes blacklist
为了构造一个自定义基因注释,我们需要用到createGeneAnnotation(),它需要你提供如下两个信息
- TxDb: 记录gene/transcript的坐标信息
- OrgDb: 用于基因名和其他基因编号的转换
继续之前的Drosophila melanogaster案例,我们需要先安装并加载相关的TxDb和OrgDb对象
if (!requireNamespace("TxDb.Dmelanogaster.UCSC.dm6.ensGene", quietly = TRUE)){
BiocManager::install("TxDb.Dmelanogaster.UCSC.dm6.ensGene")
}
if (!requireNamespace("org.Dm.eg.db", quietly = TRUE)){
BiocManager::install("org.Dm.eg.db")
}
library(TxDb.Dmelanogaster.UCSC.dm6.ensGene)
library(org.Dm.eg.db)
记着,我们构建基因注释对象
geneAnnotation <- createGeneAnnotation(TxDb = TxDb.Dmelanogaster.UCSC.dm6.ensGene, OrgDb = org.Dm.eg.db)
检查该对象,可以了解ArchR关于基因注释的存放形式
geneAnnotation
## List of length 3
## names(3): genes exons TSS
如果你没有TxDb和OrgDb对象,你也可以直接根据如下信息创建geneAnnotation对象
- "genes": GRanges对象,记录基因的坐标信息,起始位置和结束。必须要有一列是和"exons"对象中其中一列匹配
- "exons": 记录每个基因外显子的坐标。必须要有一列和"genes"对象中的一列匹配
- "GRanges": 记录TSS(转录起始位点)的坐标
geneAnnotation <- createGeneAnnotation(
TSS = geneAnnotation$TSS,
exons = geneAnnotation$exons,
genes = geneAnnotation$genes
)
1.5.2 使用非标准基因组
ArchR会做一些必要的检查,来避免你做一些ArchR觉得"不符合常理"的操作。其中就是检查你的基因组注释的seqnames都需要以"chr"开头。大部分情况下这都没有问题,除了一些基因组的染色体并不都是以"chr"作为前缀(例如玉米)。如果用ArchR分析这些基因组,你需要告知ArchR忽略染色体名前缀,否则会报错停止。你需要在创建Arrow文件前,先运行addArchRChrPrefix(chrPrefix = FALSE). 它会当前的R session里停止所有对染色体名前缀的检查操作。
此外,ArchR默认会将染色体名/seqnames转成字符串,因此如果你的seqnames都是数字,你需要以字符串形式提供这seqnames, 例如,你需要执行useSeqnames = c("1", "2", "3"),而不是useSeqnames = c(1, 2, 3)
你可以用getArchRChrPrefix随时检查当前R session是否需要染色体前缀。
1.6 创建Arrow文件
在本教程的后续部分,我们会使用Granja* et al. Nature Biotechnology 2019文章的数据进行展示,当然不是所有的数据集,我们会使用降抽样的造血细胞数据集,保证大部分人的电脑都能带动。该数据集包括了骨髓单核细胞(BMMC)和外周血单核细胞(PBMC),以及CD34+造血干细胞和骨髓前体细胞(CD34 BMMC)。
我们下载的数据以fragment格式存放,记录每个比对序列在基因组上的位置。Fragments文件是10X Genomics分析平台的其中一个输出文件,或者你也可以从BAM文件进
一旦我们得到了fragment文件,我们将它们的路径记录在一个向量中,然后作为参数传给createArrowFiles(). 在构建过程中,一些基本的元信息和矩阵会增加到各个Arrow文件中,包括TileMatrix和GeneScoreMatrix. 其中TileMarix记录的以500-bp作为分窗统计染色体上各个位置上是否有insertion(具体见addTileMatrix), GeneScoreMatrix则是基于邻近基因启动子的insetion数推断的基因表达量(见addGeneScoreMatrix())。
教程用到的数据,可以用getTutorialData进行下载,大约为0.5G。
inputFiles <- getTutorialData("Hematopoiesis")
当然这一步实际是在本地建立一个HemeFragments目录,并下载指定数据,因此如果网络太差,可以自己尝试下载。
mkdir HemeFragments && cd HemeFragments
wget 'https://jeffgranja.s3.amazonaws.com/ArchR/TestData/HemeFragments/scATAC_BMMC_R1.fragments.tsv.gz'
wget 'https://jeffgranja.s3.amazonaws.com/ArchR/TestData/HemeFragments/scATAC_CD34_BMMC_R1.fragments.tsv.gz'
wget 'https://jeffgranja.s3.amazonaws.com/ArchR/TestData/HemeFragments/scATAC_PBMC_R1.fragments.tsv.gz'
在启动项目前,我们必须得设置ArchRGenome并设置线程数threads
addArchRGenome("hg19") # hg38, mm9, mm10
addArchRThreads(threads = 16)
从现在开始,我们将会用10-15分钟的时间创建Arrow文件。对每一个样本,都会有如下操作
- 从给定文件中读取开放信息(fragments)
- 为每个细胞计算质控信息(TSS富集得分和核小体信息)
- 根据质控参数过滤细胞
- 以500bp为窗口构建全基因组范围的TileMatrix
- 使用自定义的
geneAnnotaiton创建GeneScoreMatrix, 其中geneAnnotation在我们调用addArchRGenome时定义
ArrowFiles <- createArrowFiles(
inputFiles = inputFiles,
sampleNames = names(inputFiles),
filterTSS = 4, # 这个参数不需要过高,后续可以调整
filterFrags = 1000,
addTileMat = TRUE,
addGeneScoreMat = TRUE
)
我们可以看下ArrowFiles对象,检查它是否真的是一个存放Arrow文件路径的向量
ArrowFiles
# [1] "scATAC_BMMC_R1.arrow" "scATAC_CD34_BMMC_R1.arrow"
# [3] "scATAC_PBMC_R1.arrow"
1.7 细胞质控
scATAC-seq的严格质控对于剔除低质量细胞至关重要。在ArchR中,我们考虑数据的三种信息
- 每个细胞中唯一比对数(number of unique nuclear fragments),不包括比对到线粒体的部分(unique nuclear fragments, 类似于单细胞转录组数据的表达的基因量)
- 信噪比(signal-to-background ratio)。如果是死细胞或者快死的细胞,那么DNA倾向于去染色质化,就会导致全局转座酶随机切割,体现出来就是信噪比低。
- fragment大小分布( fragment size distribution)。 由于核小体周期性,我们期望看到在147-bp附近出现一个低谷,因为缠绕一个核小体的DNA序列大约为147bp。
第一个参数, 唯一比对数非常的直观,如果一个细胞中的唯一比对太少,显然在后续分析中也没有太多可用价值,可以直接剔除掉。
第二个参数,信噪比是根据TSS富集分数进行计算。传统的混池ATAC-seq分析中,会使用TSS富集得分作为标准流程的一部分,用于确定信噪比(如 ENCODE计划)。我们和其他人通过混池ATAC-seq和scATAC-seq分析发现,TSS富集得分在大部分的细胞类型中都具有代表性。TSS富集得分的背后思想是,由于大蛋白复合体会结合在启动子区域,ATAC-seq数据更多的富集在基因的TSS区域而不基因组其他区域。通过检查这些TSS区域中心的开放水平,我们发现中心相对于两侧(两边的1900-2000 bp处)存在富集现象。因此,我们定义富集中心(TSS中心)相对于两侧区域的比值为TSS富集得分(TSS enrichment score)
传统上,我们会计算每个混池ATAC-seq样本中每个碱基的开放性,之后这个谱会被用于确定TSS富集得分。在scATAC-seq中通过该方法计算每个细胞的TSS富集得分速度较慢,并且需要很强的算力。为了提高计算效率,同时还能得到和传统计算接近的结果,我们以TSS位置为中心,以50-bp作为分窗计算两边的平均开放程度,之后该值除以TSS两侧位置(+-1900-200bp)的平均开放程度。 这种计算方法和原来相对比,两者的相关性大于0.99,并且结果几乎一样。
第三个参数fragment大小分布并不是特别重要,最好是人工检查下。由于DNA缠绕核小体的模式,我们预期在fragment大小分布中看到核小体的周期性分布。这些山峰和低谷的出现正是由于DNA缠绕0,1,2个核小体的结果(Tn5无法切割缠绕在核小体的DNA序列,只能切割两边)
ArchR默认会过滤TSS富集得分低于4或唯一比对数小于1000(也就是保留TSS富集得分大于4且唯一比对数大于1000的细胞)。切记,ArchR默认的参数最初是用于人类数据,不能直接用于其他物种。每个数据都有他的独特性,切勿生搬硬套,我们需要按照实际情况设置参数。一定要在createArrowFiles()运行前设置好该参数。
创建Arrow文件会在当前目录下生成一个"QualityControl"目录,这里面包括2个和样本相关的图。第一个图展示log10(unique nuclear fragments) vs TSS enrichment score, 虚线表示过滤阈值。第二图注释fragment大小分布图。
对我们的教程数据,我们的三个样本表现如下
BMMC:

CD34+ BMMC:

PBMC:

我们现在整理完毕我们的Arrow文件,接下来就是创建ArchRProject了。
