第12章 使用ArchR进行motif和特征富集分析
在鉴定到可靠的peak集之后,我们也会想预测有哪些转录因子参与了结合事件(binding events),从而产生了这些染色质开放位点。在分析标记peak或差异peak时,这能帮助我们更好的理解为什么某组的peak会富集某一类转录因子的结合位点。举个例子,我们想在细胞特异的染色质开放区域中找到定义谱系的关键转录因子。同样,我们也想根据其他已知特征对不同的peak进行富集分析。比如说,我们像知道是否细胞类型A的细胞特异性ATAC-seq peak对于另一组基因组区域(如ChIP-seq peak)也富集。这一章会详细介绍ArchR中的富集分析原理。
12.1 差异peak中的motif富集
继续上一章的差异peak分析,我们可以寻找在不同类型细胞富集的peak中的motif。我们需要先将motif的注释信息加入到我们的ArchRProject中。我们调用addMotifAnnotations()函数分析ArchRProject的peak中是否存在motif。运行结束后会在ArchRProject对象中加入一个新的二值矩阵,用于判断peak是否包括motif。
projHeme5 <- addMotifAnnotations(ArchRProj = projHeme5, motifSet = "cisbp", name = "Motif")
接着我们使用上一章差异检验得到的markerTest分析motif的富集情况,这是一个SummarizedExperiment对象。我们用peakAnnoEnrichments()函数分析这些差异开放peak是否富集某一类moitf。可以设置cutOff来过滤peak,例如PDR <= 0.1 & Log2FC >=0.5记录的是"Erythroid"比"Progenitor"更开放的peak。
注: peakAnnoEnrichment()能用于多种差异富集检验,在后续章节还会介绍。
motifsUp <- peakAnnoEnrichment(
seMarker = markerTest,
ArchRProj = projHeme5,
peakAnnotation = "Motif",
cutOff = "FDR <= 0.1 & Log2FC >= 0.5"
)
输出的peakAnnoEnrichment()是一个SummarizedExperiment对象,里面存放着多个assays, 记录着超几何检验的富集结果。
motifsUp
# class: SummarizedExperiment
# dim: 870 1
# metadata(0):
# assays(10): mlog10Padj mlog10p … CompareFrequency feature
# rownames(870): TFAP2B_1 TFAP2D_2 … TBX18_869 TBX22_870
# rowData names(0):
# colnames(1): Erythroid
# colData names(0):
然后,我们创建一个data.frame对象用于ggplot作图,包括motif名,矫正的p值和显著性排序。
df <- data.frame(TF = rownames(motifsUp), mlog10Padj = assay(motifsUp)[,1])
df <- df[order(df$mlog10Padj, decreasing = TRUE),]
df$rank <- seq_len(nrow(df))
正如我们所预期的那样,"Erythroid"里开放的peak富集的motif主要是GATA转录因子,符合以往研究中"GATA1"在erythroid分化中发挥的作用。
head(df)
使用ggplot展示结果,以ggrepel来标识每个TF motif名。
ggUp <- ggplot(df, aes(rank, mlog10Padj, color = mlog10Padj)) +
geom_point(size = 1) +
ggrepel::geom_label_repel(
data = df[rev(seq_len(30)), ], aes(x = rank, y = mlog10Padj, label = TF),
size = 1.5,
nudge_x = 2,
color = "black"
) + theme_ArchR() +
ylab("-log10(P-adj) Motif Enrichment") +
xlab("Rank Sorted TFs Enriched") +
scale_color_gradientn(colors = paletteContinuous(set = "comet"))
ggUp

通过设置Log2FC <= 0.5我们可以挑选出在"Progenitor"里更加开放的peak,然后分析其中富集的motif。
motifsDo <- peakAnnoEnrichment(
seMarker = markerTest,
ArchRProj = projHeme5,
peakAnnotation = "Motif",
cutOff = "FDR <= 0.1 & Log2FC <= -0.5"
)
motifsDo
准备绘图所需数据框
df <- data.frame(TF = rownames(motifsDo), mlog10Padj = assay(motifsDo)[,1])
df <- df[order(df$mlog10Padj, decreasing = TRUE),]
df$rank <- seq_len(nrow(df))
此时,我们会发现在"Progenitor"细胞更加开放的peak中,更多富集RUNX, ELF和CBFB。
head(df)
# TF mlog10Padj rank
# 326 ELF2_326 88.68056 1
# 733 RUNX1_733 64.00586 2
# 801 CBFB_801 53.55426 3
# 732 RUNX2_732 53.14766 4
# 734 ENSG00000250096_734 53.14766 5
# 336 SPIB_336 52.79666 6
使用ggplot展示结果。
ggDo <- ggplot(df, aes(rank, mlog10Padj, color = mlog10Padj)) +
geom_point(size = 1) +
ggrepel::geom_label_repel(
data = df[rev(seq_len(30)), ], aes(x = rank, y = mlog10Padj, label = TF),
size = 1.5,
nudge_x = 2,
color = "black"
) + theme_ArchR() +
ylab("-log10(FDR) Motif Enrichment") +
xlab("Rank Sorted TFs Enriched") +
scale_color_gradientn(colors = paletteContinuous(set = "comet"))
ggDo
plotFDF()函数能够以可编辑的矢量版本保存图片。
plotPDF(ggUp, ggDo, name = "Erythroid-vs-Progenitor-Markers-Motifs-Enriched", width = 5, height = 5, ArchRProj = projHeme5, addDOC = FALSE)
12.2 标记Peak的motif富集分析
和之前利用差异peak的motif富集分析类似,我们同样能用getMarkerFeatures()分析标记peak里富集的motif。
我们向函数peakAnnotationEnrichment()传入存放标记peak的SummarizedExperiment对象,即markersPeaks
enrichMotifs <- peakAnnoEnrichment(
seMarker = markersPeaks,
ArchRProj = projHeme5,
peakAnnotation = "Motif",
cutOff = "FDR <= 0.1 & Log2FC >= 0.5"
)
输出的peakAnnoEnrichment()是一个SummarizedExperiment对象,里面存放着多个assays, 记录着超几何检验的富集结果。
enrichMotifs
# class: SummarizedExperiment
# dim: 870 11
# metadata(0):
# assays(10): mlog10Padj mlog10p … CompareFrequency feature
# rownames(870): TFAP2B_1 TFAP2D_2 … TBX18_869 TBX22_870
# rowData names(0):
# colnames(11): B CD4.M … PreB Progenitor
# colData names(0):
直接用plotEnrichHeatmap()函数绘制不同细胞组的富集的motif。通过设置参数n限制每个细胞分组中展示的motif。
heatmapEM <- plotEnrichHeatmap(enrichMotifs, n = 7, transpose = TRUE)
使用ComplexHeatmap::draw()函数展示结果
ComplexHeatmap::draw(heatmapEM, heatmap_legend_side = "bot", annotation_legend_side = "bot")

plotFDF()函数能够以可编辑的矢量版本保存图片。
plotPDF(heatmapEM, name = "Motifs-Enriched-Marker-Heatmap", width = 8, height = 6, ArchRProj = projHeme5, addDOC = FALSE)
12.3 ArchR富集分析
除了分析peak中富集motif, ArchR还能进行个性化的富集分析。为了方便这类数据探索,我们已人工确定了一些特征数据集,它们能比较容易地在我们感兴趣的peak区间进行检验。我们接下来将会逐个介绍这些特征数据集。该分析最初受LOLA启发。
12.3.1 Encode TF 结合位点
ENCODE协会已经将TF结合位点(TFBS)匹配到多种细胞类型和因子中。我们可以利用这些TFBS去更好地理解聚类结果。例如,我们可以根据富集结果去判断未知细胞类型的可能类型。为了能够使用ENCODE TFBS特征集进行分析,我们需要调用addArchRAnnotations()函数,设置collection = "EncodeTFBS". 和使用addPeakAnnotations()类似,这会创建一个二值矩阵,记录我们的标记peak是否和ENCODE TFBS有重叠。
projHeme5 <- addArchRAnnotations(ArchRProj = projHeme5, collection = "EncodeTFBS")
我们接着使用peakAnnoEnrichment()函数分析这些 ENCODE TFBS是否在我们的peak中富集。
enrichEncode <- peakAnnoEnrichment(
seMarker = markersPeaks,
ArchRProj = projHeme5,
peakAnnotation = "EncodeTFBS",
cutOff = "FDR <= 0.1 & Log2FC >= 0.5"
)
和之前一样,该函数返回一个SummarizedExperiment对象。
enrichEncode
# class: SummarizedExperiment
# dim: 689 11
# metadata(0):
# assays(10): mlog10Padj mlog10p … CompareFrequency feature
# rownames(689): 1.CTCF-Dnd41… 2.EZH2_39-Dnd41… …
# 688.CTCF-WERI_Rb_1… 689.CTCF-WI_38…
# rowData names(0):
# colnames(11): B CD4.M … PreB Progenitor
# colData names(0):
我们可以使用plotEnrichHeatmap函数从富集结果中创建热图。
heatmapEncode <- plotEnrichHeatmap(enrichEncode, n = 7, transpose = TRUE)
然后用ComplexHeatmap::draw()绘制热图
ComplexHeatmap::draw(heatmapEncode, heatmap_legend_side = "bot", annotation_legend_side = "bot")

plotFDF()函数能够以可编辑的矢量版本保存图片。
plotPDF(heatmapEncode, name = "EncodeTFBS-Enriched-Marker-Heatmap", width = 8, height = 6, ArchRProj = projHeme5, addDOC = FALSE)
12.3.2 混池ATAC-seq
和ENCODE TFBS类似,我们还可以使用混池ATAC-seq实验鉴定的peak,分析两者的重叠情况。通过设置collection="ATAC"来调用混池ATAC-seqpeak数据集。
projHeme5 <- addArchRAnnotations(ArchRProj = projHeme5, collection = "ATAC")
接着通过设置peakAnnotation = "ATAC"检验我们的标记peak是否富集了混池ATAC-seq的peak。
enrichATAC <- peakAnnoEnrichment(
seMarker = markersPeaks,
ArchRProj = projHeme5,
peakAnnotation = "ATAC",
cutOff = "FDR <= 0.1 & Log2FC >= 0.5"
)
和之前一样,该函数会输出SummarizedExperiment对象,记录着富集结果
enrichATAC
# class: SummarizedExperiment
# dim: 96 11
# metadata(0):
# assays(10): mlog10Padj mlog10p … CompareFrequency feature
# rownames(96): Brain_Astrocytes Brain_Excitatory_neurons … Heme_MPP
# Heme_NK
# rowData names(0):
# colnames(11): B CD4.M … PreB Progenitor
# colData names(0):
我们用plotEnrichHeatmap()函数基于SummarizedExperiment绘制富集热图
heatmapATAC <- plotEnrichHeatmap(enrichATAC, n = 7, transpose = TRUE)
使用ComplexHeatmap::draw()绘制结果
ComplexHeatmap::draw(heatmapATAC, heatmap_legend_side = "bot", annotation_legend_side = "bot")

plotFDF()函数能够以可编辑的矢量版本保存图片。
plotPDF(heatmapATAC, name = "ATAC-Enriched-Marker-Heatmap", width = 8, height = 6, ArchRProj = projHeme5, addDOC = FALSE)
12.3.3 Codex TFBS
相同类型的分析还能用于 CODEX TFBS,只要设置collection = "Codex"即可
projHeme5 <- addArchRAnnotations(ArchRProj = projHeme5, collection = "Codex")
enrichCodex <- peakAnnoEnrichment(
seMarker = markersPeaks,
ArchRProj = projHeme5,
peakAnnotation = "Codex",
cutOff = "FDR <= 0.1 & Log2FC >= 0.5"
)
heatmapCodex <- plotEnrichHeatmap(enrichCodex, n = 7, transpose = TRUE)
ComplexHeatmap::draw(heatmapCodex, heatmap_legend_side = "bot", annotation_legend_side = "bot")

于是我们就保存图片了
plotPDF(heatmapCodex, name = "Codex-Enriched-Marker-Heatmap", width = 8, height = 6, ArchRProj = projHeme5, addDOC = FALSE)
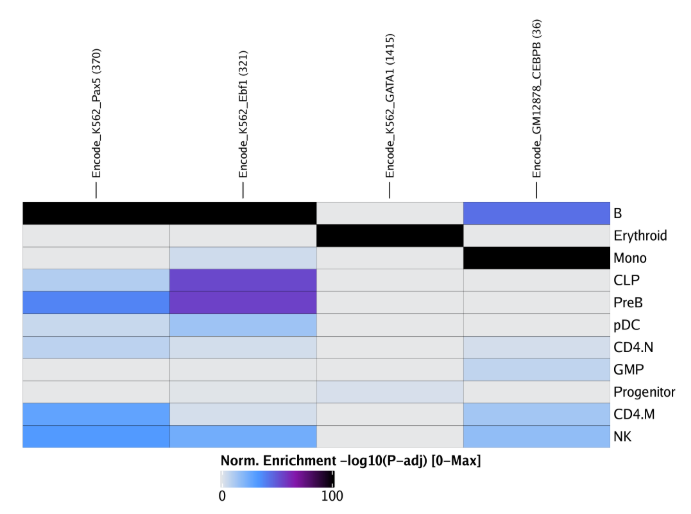
12.4 自定义富集
除了之前这些经过人工审核的注释数据集,ArchR还能处理用户自定义注释信息来执行富集分析。接下来,我们会介绍如何根据ENCODE ChIP-seq实验来创建自定义的注释信息。
首先,我们先提供后续将被使用并下载的数据集,也可以提供本地文件。
EncodePeaks <- c(
Encode_K562_GATA1 = "https://www.encodeproject.org/files/ENCFF632NQI/@@download/ENCFF632NQI.bed.gz",
Encode_GM12878_CEBPB = "https://www.encodeproject.org/files/ENCFF761MGJ/@@download/ENCFF761MGJ.bed.gz",
Encode_K562_Ebf1 = "https://www.encodeproject.org/files/ENCFF868VSY/@@download/ENCFF868VSY.bed.gz",
Encode_K562_Pax5 = "https://www.encodeproject.org/files/ENCFF339KUO/@@download/ENCFF339KUO.bed.gz"
)
然后,我们用addPeakAnnotation()函数在ArchRProject函数中增加自定义注释。我们这里将其命名为"ChIP"
projHeme5 <- addPeakAnnotations(ArchRProj = projHeme5, regions = EncodePeaks, name = "ChIP")
和之前一样,我们使用peakAnnoEnrichment()函数根据自定义的注释信息执行peak注释富集分析
enrichRegions <- peakAnnoEnrichment(
seMarker = markersPeaks,
ArchRProj = projHeme5,
peakAnnotation = "ChIP",
cutOff = "FDR <= 0.1 & Log2FC >= 0.5"
)
并以相同的步骤生成注释热图。
heatmapRegions <- plotEnrichHeatmap(enrichRegions, n = 7, transpose = TRUE)
ComplexHeatmap::draw(heatmapRegions, heatmap_legend_side = "bot", annotation_legend_side = "bot")

plotFDF()函数能够以可编辑的矢量版本保存图片。
plotPDF(heatmapRegions, name = "Regions-Enriched-Marker-Heatmap", width = 8, height = 6, ArchRProj = projHeme5, addDOC = FALSE)
