目前一些生信工具,例如samtools,gffread 是用C/C++编写的,他们在编码染色体的坐标时,用的一般是int32,对应数值范围是-2,147,483,648 ~2,147,483,647。在计算机课上,有一个知识点,叫做整型溢出,也就是如果你要编码的数字超过编码范围,那他就会从最小开始,也就是2,147,483,647的下一位数字是-2,147,483,648。
举个例子,如果你的染色体比较长,超过了500mb,那么 samtools index建立索引时,会报错 Region 536761809..536937545 cannot be stored in a bai index. Try using a csi index: Numerical result out of range 。虽然说报错建议你使用csvi,确实能解决了一些问题。然而,如果你的染色体离谱的大,那么csi也救不了你,他还是会报错 Unsorted positions on sequence #1: 2147481987 followed by -2147483162。对于gffread,它就更加离谱了,他不会报错,因为他直接将错就错,也就是你经过gffread转换后的gtf/gff,会在结果中看到负数的坐标。
其他还有一些软件,例如bwa, plink可能都不支持那么长的染色体。 STAR虽然能够建立索引,也能回帖,但是你会发现得到的BAM文件,无人能处理。还有, NCBI上会有染色体的长度限制,超过这一长度需要你进行拆分。
同时,考虑, 目前的大基因组,都不可能是T2T基因组,也不可能有一个contig长度会超过500 mb,于是我写了一个脚本,他的逻辑就是把这些scaffolding的基因组,打碎成不那么连续的部分,然后基于这个坐标生成新的GTF或者GFF,代码见 https://github.com/xuzhougeng/myscripts/blob/master/misc/splite_super_big_genome.py
同时,我把代码打包成了一个TBtools的插件,插件和测试数据放在了一起。使用方法非常简单,就是选择你的fasta的路径,你的gff文件路径,设置输出路径就行了,举个例子
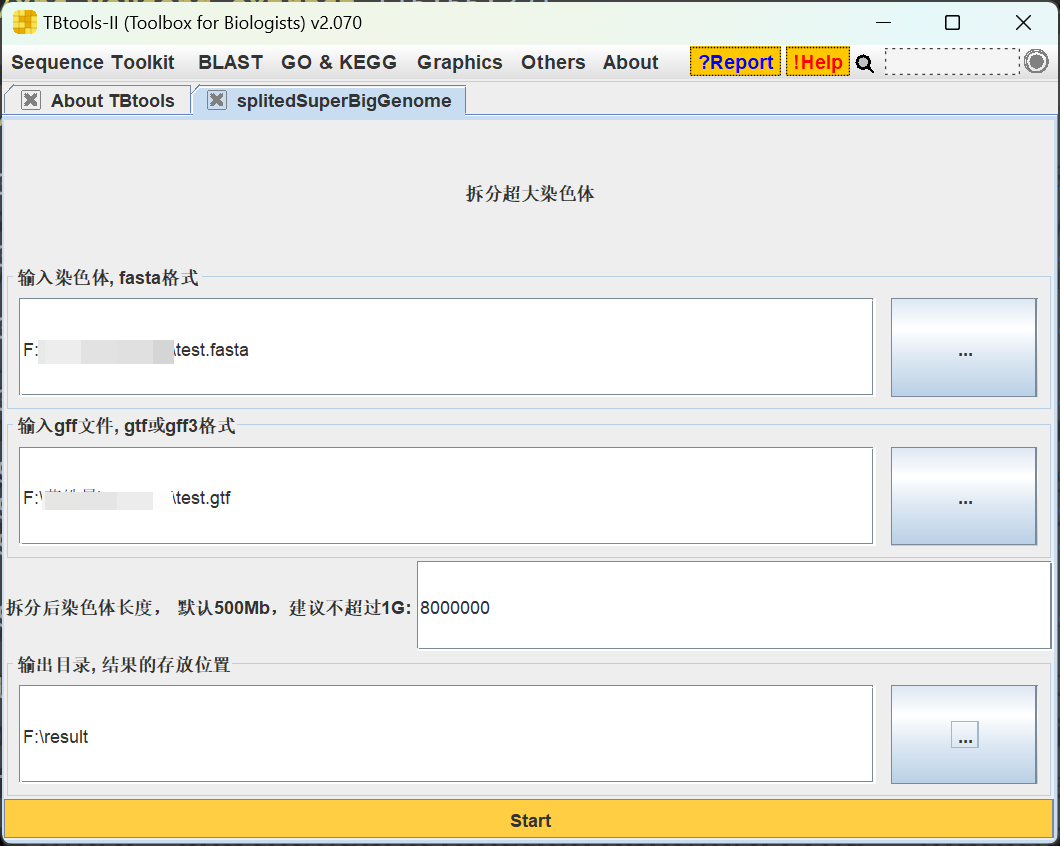
测试数据地址: 链接:https://pan.baidu.com/s/15ixljZVarc6udOSS0CCH6w?pwd=zgnh 提取码:zgnh
注意插件的使用,需要安装python环境,可以参考 https://www.yuque.com/cjchen/hirv8i/xpffndywtchwg83z 进行安装。
参考资料
- TBtools文档: https://www.yuque.com/cjchen/hirv8i/xq65ml
