binmapr目前代码正在重构,目前无法从CRAN进行下载,本篇教程仅为存档用。
binmapr是我折腾的一个R包,它能够将NGS测序得到SNP数据基于binmap进行纠错,用于更好的遗传定位。
在阅读本文之前,请先花点时间看看Bin, Bin, Bin!Map, Map, Map Now!, 我只是将里面的步骤整理成R包方便调用而已。
首先你得安装并加载R包。因为这个R包目前主要是方便自己使用,所以托管在GitHub上,需要用devtools进行安装
devtools::install_github("xuzhougeng/binmapr")
之后就可以和其他R包一样正常使用
library(binmapr)
R包的使用非常简单,就是调用batchCallGeno 将原本的genotype矩阵按照15bp对snp进行纠正
geno <- batchCallGeno(GT_flt, CHROM = CHROM,
outdir = ".",
pos.start = 7, fix.size = 10)
因此,你只要提供一个符合要求的输入即可。
以李广伟师兄的数据为例,我已经将其整理成示例数据,因此可以可以通过下面两行命令读取
data(geno)
data(pheno)
这两个都是数据框,其中geno存放的是基因型数据,而pheno存放的是表型数据
为了能够让batchCallGeno运行,我们需要将geno数据转成一个矩阵,其中行名是位置信息,列名是样本信息
GT <- as.matrix(geno[,-1:-4])
row.names(GT) <- paste0(geno$CHR, "_", geno$POS)
由于每个位点都在所有样本中都不一定存在,因此可以考虑过滤一些缺失比较多的位点.
miss_ratio <- rowSums(is.na(GT)) / ncol(GT)
GT_flt <- GT[miss_ratio < 0.20, ]
我这里过滤掉缺失大于20%的位点,原本是打算用5%,未曾想到这个标准过滤下去,90%的数据都快没了,吓得我赶紧用summary(miss_ratio)看了一波分布,改了下标准。
有了合适的数据类型后,就可以调用batchCallGeno函数了。运行结束后,除了得到一个列表外存放基因型外,还会在当前目录下输出csv和pdf文件。其中csv是表型数据,而pdf则是纠错前后的基因型分布。
CHROM <- unique(geno$CHR)
geno_out <- batchCallGeno(GT_flt, CHROM = CHROM,
pos.start = 7, fix.size = 10)
介绍下几个参数
CHROM: 用于构建binmap的染色体pos.start, 该参数用于绘图时从行名中提取坐标,例如Chr1_1245需要设置为pos.start=6, 这里命名为chr01_12345所以要设置为pos.start=7fix.size, 它和纠错有关,比如说你的基因型是00000100000, 中间的1很可能是错误的,因为它只出现了一次。fix.size需要根据具体数据来调整。
下一步,我们就可以用方差分析的方法来寻找和表型相关的区间
# Load phenotype
anova_analysis <- function(x, phenotype){
p.lmout <- lm(phenotype ~ x, na.action = na.exclude)
p <- anova(p.lmout)[,5][1]
return(p)
}
geno_mt <- geno_out[[1]]
geno_mt_reorder <- geno_mt[,pheno$ID]
pvals <- apply(geno_mt_reorder,
1,
anova_analysis,
phenotype = pheno$PH)
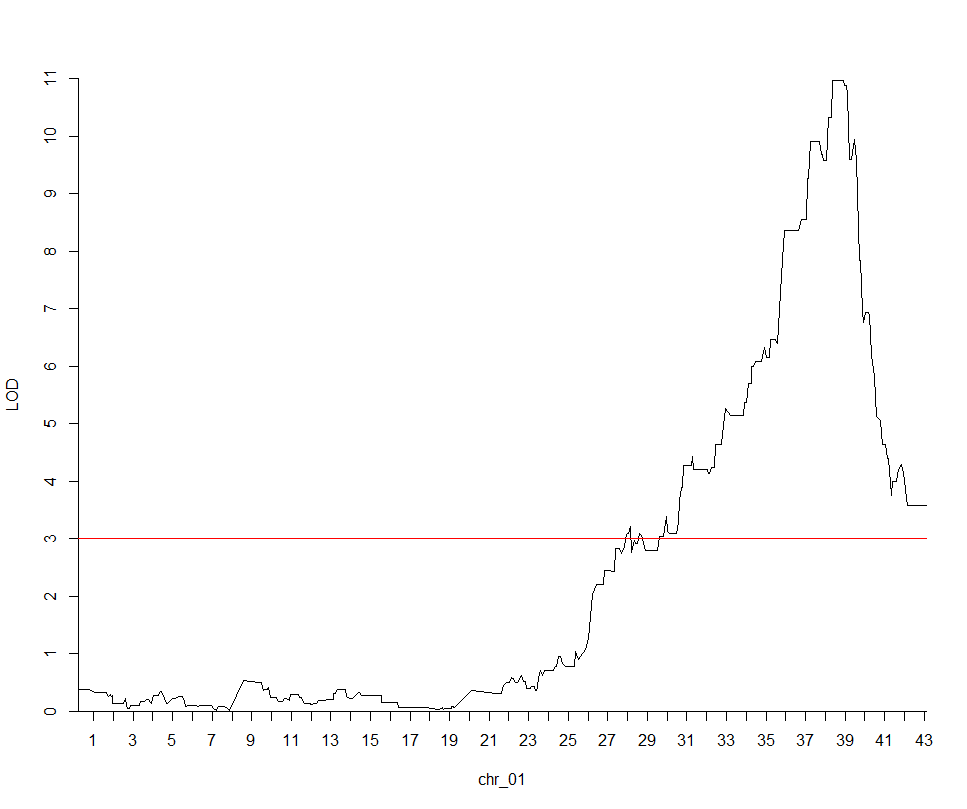
接着画图
pos <- as.numeric(substring(row.names(geno_mt_reorder), 7))
plotQtl(pos = pos,
pvalue = pvals,
chr.name = "chr_01",
ymax = 11,
threshold = 3)

我们可以从中看到一个非常显著的峰,里面的基因中就有一个碰巧是水稻里的绿色革命基因,sd1 LOC_Os01g66100 物理区间是 38382382-38385504,距QTL最显著位置只有258kb的距离,对于一个只有172个个体的F2群体而言,结果是不是已经很不错了。
