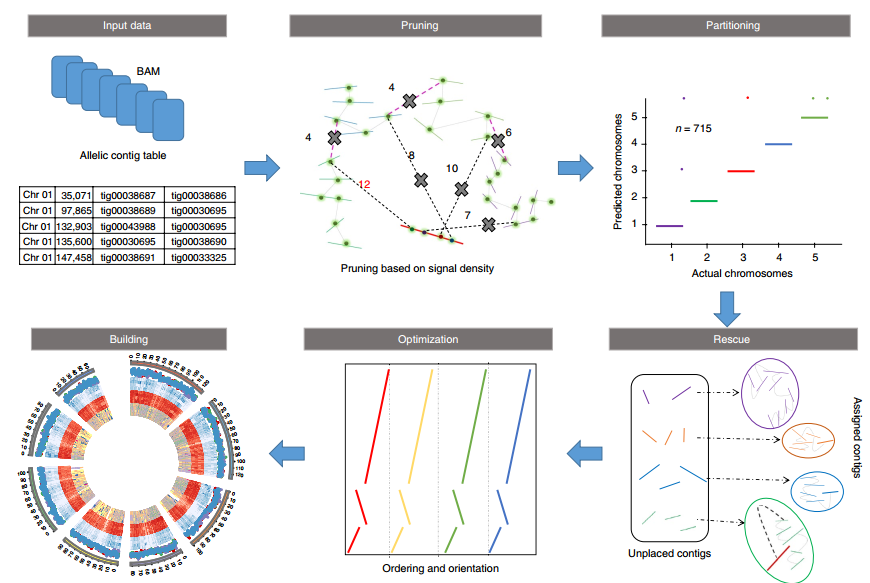
ALLHiC: 辅助组装简单的二倍体基因组
ALLHiC除了能够解决复杂基因组(高杂合,多倍体)的基因组组装问题,还能够搞定简单的基因组组装,毕竟本质上都是利用HiC的信号。
ALLHiC大概有两个优势:
- 对同源多倍体是一种解决方案
- 当contig组装比较短的时候,大部分情况allhic的排序优于其他软件(如果contig很长的话,可以优先考虑下3d-dna打断,过长的contig或许是错误组装造成)
参考使用ALLHiC基于HiC数据辅助基因组组装完成软件安装和前三步的数据预处理,将Fastq文件转成BAM文件。
下面假定你的初步组装结果是draft.asm.fasta, Fastq预处理后的文件是sample.clean.bam
之后,我们将contig根据相互之间的信号强弱关系进行分组,至于分成多少组要根据物种具体的染色体数目而定。
ALLHiC_partition -b sample.clean.bam -r draft.asm.fasta -e AAGCTT -k 16
# -k: 聚类组数
# -e: 酶切类型, 预设HindIII: AAGCTT; MboI: GATC
# -m: 每个contig最少的酶切位点, 默认25
要根据自己的实际建库方法选择酶切位点的识别序列,注, DpnI, DpnII, MboI, Sau3AI 这些酶都是识别相同的序列,仅仅是对甲基化敏感度不同。
接着,提取CLM和每个contig的酶切数
allhic extract sample.clean.bam draft.asm.fasta --RE AAGCTT
然后是对每个组的contig位置和方向进行优化
# 如下命令可以通过循环同时运行
allhic optimize sample.clean.counts_AAGCTT.16g1.txt sample.clean.clm
allhic optimize sample.clean.counts_AAGCTT.16g2.txt sample.clean.clm
...
allhic optimize sample.clean.counts_AAGCTT.16g16.txt sample.clean.clm
最终获取染色体级别的组装
ALLHiC_build draft.asm.fasta
绘制热图对组装进行评估
# 获取染色体长度
perl getFaLen.pl -i groups.asm.fasta -o len.txt
# 构建chrn.list
grep 'merge.clean.counts_GATC' len.txt > chrn.list
# 绘图, 500k表示分辨率
ALLHiC_plot sample.clean.bam groups.agp chrn.list 500k pdf
其中getFaLen.pl可以从https://github.com/tangerzhang/my_script获取
