本教程借鉴https://github.com/tanghaibao/jcvi/wiki/MCscan-(Python-version).
我们先从http://plants.ensembl.org/index.html选择两个物种做分析, 这里选择的就是前两个物种,也就是拟南芥和水稻(得亏没有小麦和玉米)

我们下载它的GFF文件,cdna序列和蛋白序列
#Athaliana
wget ftp://ftp.ensemblgenomes.org/pub/plants/release-44/fasta/arabidopsis_thaliana/cdna/Arabidopsis_thaliana.TAIR10.cdna.all.fa.gz
wget ftp://ftp.ensemblgenomes.org/pub/plants/release-44/fasta/arabidopsis_thaliana/pep/Arabidopsis_thaliana.TAIR10.pep.all.fa.gz
wget ftp://ftp.ensemblgenomes.org/pub/plants/release-44/gff3/arabidopsis_thaliana/Arabidopsis_thaliana.TAIR10.44.gff3.gz
#Osativa
wget ftp://ftp.ensemblgenomes.org/pub/plants/release-44/fasta/oryza_sativa/cdna/Oryza_sativa.IRGSP-1.0.cdna.all.fa.gz
wget ftp://ftp.ensemblgenomes.org/pub/plants/release-44/fasta/oryza_sativa/pep/Oryza_sativa.IRGSP-1.0.pep.all.fa.gz
wget ftp://ftp.ensemblgenomes.org/pub/plants/release-44/gff3/oryza_sativa/Oryza_sativa.IRGSP-1.0.44.gff3.gz
保证要有6个文件以便下游分析
$ ls
Arabidopsis_thaliana.TAIR10.44.gff3.gz Arabidopsis_thaliana.TAIR10.pep.all.fa.gz Oryza_sativa.IRGSP-1.0.cdna.all.fa.gz
Arabidopsis_thaliana.TAIR10.cdna.all.fa.gz Oryza_sativa.IRGSP-1.0.44.gff3.gz Oryza_sativa.IRGSP-1.0.pep.all.fa.gz
我们分析只需要用到每个基因最长的转录本就行,之前我用的是自己写的脚本,但其实我发现jcvi其实可以做到这件事情
先将gff转成bed格式,
python -m jcvi.formats.gff bed --type=mRNA --key=transcript_id Arabidopsis_thaliana.TAIR10.44.gff3.gz > ath.bed
python -m jcvi.formats.gff bed --type=mRNA --key=transcript_id Oryza_sativa.IRGSP-1.0.44.gff3.gz > osa.bed
然后将bed进行去重复
python -m jcvi.formats.bed uniq ath.bed
python -m jcvi.formats.bed uniq osa.bed
最后我们得到了ath.uniq.bed和osa.uniq.bed, 根据bed文件第4列就可以用于提取cds序列和蛋白序列。
# Athaliana
seqkit grep -f <(cut -f 4 ath.uniq.bed ) Arabidopsis_thaliana.TAIR10.cdna.all.fa.gz | seqkit seq -i > ath.cds
seqkit grep -f <(cut -f 4 ath.uniq.bed ) Arabidopsis_thaliana.TAIR10.pep.all.fa.gz | seqkit seq -i > ath.pep
# Osativa
seqkit grep -f <(cut -f 4 osa.uniq.bed ) Oryza_sativa.IRGSP-1.0.cdna.all.fa.gz | seqkit seq -i > osa.cds
seqkit grep -f <(cut -f 4 osa.uniq.bed ) Oryza_sativa.IRGSP-1.0.pep.all.fa.gz | seqkit seq -i > osa.pep
这里用到的seqkit建议学习,非常好用
下面使用python -m jcvi.compara.catalog ortholog进行共线性分析,这是一个非常行云流水的过程(除非你报错)
新建一个文件夹,方便在报错的时候,把全部都给删了,
mkdir -p cds && cd cds
ln -s ../ath.cds ath.cds
ln -s ../ath.uniq.bed ath.bed
ln -s ../osa.cds osa.cds
ln -s ../osa.uniq.bed osa.bed
运行代码
python -m jcvi.compara.catalog ortholog --no_strip_names ath osa
输出结果如下
$ ls ath.osa.*
ath.osa.anchors ath.osa.last ath.osa.last.filtered ath.osa.lifted.anchors ath.osa.pdf
其中我们最感兴趣都是pdf结果,不出意外没啥共线性。

我们还可以用蛋白序列做共线性分析
# 在之前输出cds,pep都文件夹操作
mkdir -p pep && cd pep
ln -s ../ath.pep ath.pep
ln -s ../ath.uniq.bed ath.bed
ln -s ../osa.pep osa.pep
ln -s ../osa.uniq.bed osa.bed
运行代码
python -m jcvi.compara.catalog ortholog --dbtype prot --no_strip_names ath osa
我之前以为他不可以基于蛋白序列分析,幸亏有人提醒。

你会发现这是一个自动化分析流程,我们只是提供了4个文件,它就完成了一些列事情。它生成的文件里除了PDF外,其他还有啥用呢?
- ath.osa.last: 基于LAST的比对结果
- ath.osa.last.filtered: LAST的比对结果过滤串联重复和低分比对
- ath.osa.anchors: 高质量的共线性区块
- ath.osa.lifted.anchors:增加了额外的锚点,形成最终的共线性区块
anchors文件特别有用,之后会写一篇介绍如何利用他进行可视化,这里介绍它的格式。
###
AT1G28395.5 Os01t0238800-02 66
AT1G28440.1 Os01t0239700-02 1360
AT1G28480.1 Os01t0241400-01 136
AT1G28510.1 Os01t0242300-01 241
###
AT1G11100.3 Os01t0779400-01 943
AT1G11125.1 Os01t0779800-01 52
AT1G11160.2 Os01t0780400-02 535
AT1G11180.1 Os01t0780500-01 483
AT1G11330.2 Os01t0784700-00 742
AT1G11360.1 Os01t0783500-01 305
AT1G11540.2 Os01t0786800-01 422
AT1G11570.3 Os01t0788200-01 162
AT1G11580.2 Os01t0788400-01 550
AT1G11630.1 Os01t0793200-01 321
每个共线性区块以###进行分隔, 第一列是检索的基因,第二列是被检索的基因,第三列则是两个序列的BLAST的bit score,值越大可靠性越高。
用水稻和拟南芥进行了比较之后,发现后面基本上也没啥可以分析了。因此下面基于「JCVI教程」如何基于物种的CDS的blast结果绘制点图(dotplot)得到的cds和bed文件进行分析。
之前已经得到了如下四个文件
ls ???.???
aly.bed aly.cds ath.bed ath.cds
所以我们只要运行
python -m jcvi.compara.catalog ortholog --no_strip_names aly ath
就得到了一个非常好看的点图

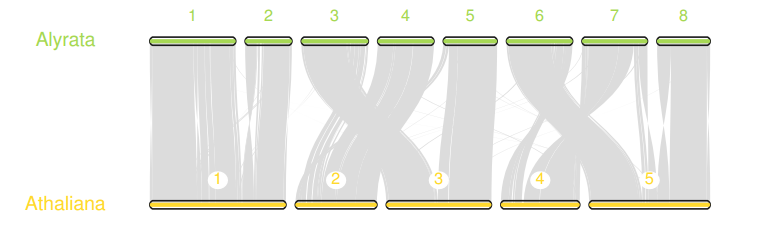
我们可以发现,都作为Arabidopsis属的两个物种,他们之间存在很高的同源性,并且同源区比例是1:1,

这其实和2011年的Nature Genetics上Alyrata的文章的结果是相似的,只不过他不是用点图进行展示

我们也可以用JCVI的画图模块实现这种效果,只不过还需要一点额外操作,创建如下三个文件
- seqids: 需要展现哪些序列
- layout: 不同物种的在图上的位置
- .simple: 从
.anchors文件创建的更简化格式
第一步,创建.simple文件
python -m jcvi.compara.synteny screen --minspan=30 --simple aly.ath.anchors aly.ath.anchors.new
第二步, 创建seqid文件,非常简单,就是需要展示的scaffold或染色体的编号
scaffold_1,scaffold_2,scaffold_3,scaffold_4,scaffold_5,scaffold_6,scaffold_7,scaffold_8
Chr1,Chr2,Chr3,Chr4,Chr5
第二步,创建layout文件,用于设置绘制的一些选项。
# y, xstart, xend, rotation, color, label, va, bed
.6, .2, .8, 0, , Alyrata, top, aly.bed
.4, .2, .8, 0, , Athaliana, top, ath.bed
# edges
e, 0, 1, aly.ath.anchors.simple
注意, #edges下的每一行开头都不能有空格
最后运行下面的命令,会得到一个karyotype.pdf
python -m jcvi.graphics.karyotype seqids layout

如何让这个图垂直呢?(导入AI里就好了)
