第8章: 使用scRNA-seq定义cluster类型
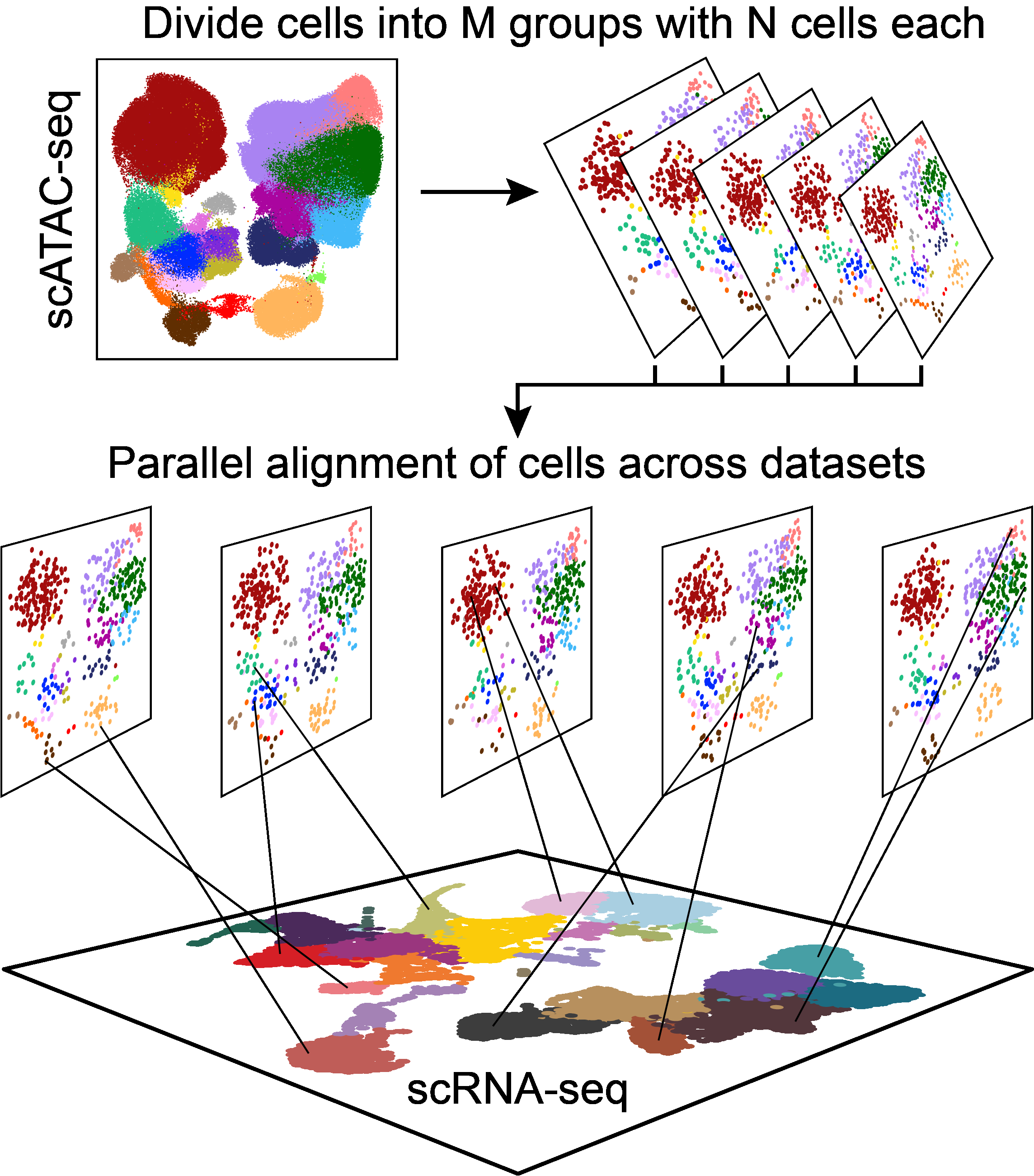
除了使用基因得分定义细胞类群以外,ArchR还能整合scRNA-seq数据。通过将scATAC-seq数据里的基因得分矩阵和scRNA-seq数据的基因表达量矩阵进行对比,ArchR就能将scATAC-seq的细胞比对到scRNA-seq的细胞,实现两种数据的整合。之后,我们借助scRNA-seq数据已经定义的细胞类群,或者整合后的scRNA-seq的基因表达量来注释细胞类群。在代码内部,我们调用了Seurat::FindTransferAnchors(),从而实现两种数据集之间的比较。当然,ArchR不是简单地对函数进行封装,而是在此基础上通过对数据的拆分,实现并行计算,从而能将该流程应用到更大规模的细胞中。

该整合过程实际上会寻找scATAC-seq和scRNA-seq两者中最相似的细胞,然后将scRNA-seq中对应的细胞表达量赋值给scATAAC-seq细胞。最后,scATAC-seq中每个细胞都会有基因表达量特征,能被用于下游分析。这一章会阐述如何利用该信息定义细胞类型,之后会介绍如何使用连接的scRNA-seq数据做更加复杂的分析,例如识别预测的顺式调控元件。我们相信由于多组学单细胞谱的商业化,这类整合分析将会越来越多。同时,在ArchR中使用公共数据里匹配细胞类型的scRNA-seq数据或者自己使用目标样本得到的scRNA-seq数据也能加强scATAC-seq分析。
8.1 scATAC-seq细胞和scRNA-seq细胞跨平台连接
为了能将我们教程中的scATAC-seq数据与其匹配的scRNA-seq数据进行整合,我们将使用 Granja* et al (2019) 里的造血细胞scRNA-seq数据。
scRNA-seq数据以 RangedSummarizedExperiment对象保存,大小为111MB。此外,ArchR还接受未经修改的Seurat对象作为整合流程的输入。我们使用download.file下载数据
if(!file.exists("scRNA-Hematopoiesis-Granja-2019.rds")){
download.file(
url = "https://jeffgranja.s3.amazonaws.com/ArchR/TestData/scRNA-Hematopoiesis-Granja-2019.rds",
destfile = "scRNA-Hematopoiesis-Granja-2019.rds"
)
}
下载之后,我们使用readRDS进行读取,并查看该对象
seRNA <- readRDS("scRNA-Hematopoiesis-Granja-2019.rds")
seRNA
## class: RangedSummarizedExperiment
## dim: 20287 35582
## metadata(0):
## assays(1): counts
## rownames(20287): FAM138A OR4F5 … S100B PRMT2
## rowData names(3): gene_name gene_id exonLength
## colnames(35582): CD34_32_R5:AAACCTGAGTATCGAA-1
## CD34_32_R5:AAACCTGAGTCGTTTG-1 …
## BMMC_10x_GREENLEAF_REP2:TTTGTTGCATGTGTCA-1
## BMMC_10x_GREENLEAF_REP2:TTTGTTGCATTGAAAG-1
## colData names(10): Group nUMI_pre … BioClassification Barcode
从输出信息中,我们可以发现它里面有基因表达量的count矩阵和对应的元信息。
元信息列里的BioClassification记录着scRNA-seq数据中每个细胞对应的细胞类型分类
colnames(colData(seRNA))
# [1] "Group" "nUMI_pre" "nUMI"
# [4] "nGene" "initialClusters" "UMAP1"
# [7] "UMAP2" "Clusters" "BioClassification"
# [10] "Barcode"
使用table(),我们可以看到scRNA-seq细胞类型每一群的细胞数
table(colData(seRNA)$BioClassification)
# 01_HSC 02_Early.Eryth 03_Late.Eryth 04_Early.Baso 05_CMP.LMPP
# 1425 1653 446 111 2260
# 06_CLP.1 07_GMP 08_GMP.Neut 09_pDC 10_cDC
# 903 2097 1050 544 325
#11_CD14.Mono.1 12_CD14.Mono.2 13_CD16.Mono 14_Unk 15_CLP.2
# 1800 4222 292 520 377
# 16_Pre.B 17_B 18_Plasma 19_CD8.N 20_CD4.N1
# 710 1711 62 1521 2470
# 21_CD4.N2 22_CD4.M 23_CD8.EM 24_CD8.CM 25_NK
# 2364 3539 796 2080 2143
# 26_Unk
# 161
我们后续会用到两种整合方法。第一种是无约束整合,我们对scATAC-seq实验里的细胞不作任何假设,直接尝试将这些细胞和scRNA-seq实验里的任意细胞进行配对。这是一种初步可行方案,后续会根据这一步得到的结果,对整合步骤进行约束来提升跨平台配对的质量。第二种方法是约束整合,即利用对细胞类型的先验知识限制搜索范围。举个例子,如果我们知道scATAC-seq中的Cluster A,B,C对应着三种不同的T细胞,scRNA-seq中的Cluster X,Y对应着两种不同的T细胞,我们告诉ArchR只需要尝试将scATAC-seq中的Cluster A,B,C跟scRNA-seq中的Cluster X,Y进行配对。下面,我们将先以无约束整合初步地鉴定每一种聚类的类型,然后根据分析结果做更加细致的约束整合。
8.1.1 无约束整合
我们使用addGeneIntegrationMatrix()对scATAC-seq和scRNA-seq数据进行整合。正如之前所提到的,该函数的seRNA参数接受Seurat或RangedSummarizedExperiment对象作为输入。因为第一轮是探索性质的无约束整合,因此,我们不会将结果保存在Arrow文件中(addToArrow=FALSE)。整合后的矩阵将会根据matrixName进行命名,存放在ArchRProject中。该函数的其他参数对应cellColData中列名用于存放额外的信息,nameCell存放scRNA-seq中匹配的细胞ID,nameGroup存放scRNA-seq细胞中的分组ID,nameScore存放跨平台整合得分。
projHeme2 <- addGeneIntegrationMatrix(
ArchRProj = projHeme2,
useMatrix = "GeneScoreMatrix",
matrixName = "GeneIntegrationMatrix",
reducedDims = "IterativeLSI",
seRNA = seRNA,
addToArrow = FALSE,
groupRNA = "BioClassification",
nameCell = "predictedCell_Un",
nameGroup = "predictedGroup_Un",
nameScore = "predictedScore_Un"
)
无约束整合的结果可能并不准确,但是为后续更加精细的约束分析奠定了基础。
8.1.2 约束整合
现在我们已经有了初步的无约束整合结果,我们就有了大致的细胞类型分布情况,接着就是优化整合结果。
因为我们教程里的数据来自于造血细胞,我们将会在理想状态下将类似的细胞整合在一起。首先,我们先确认scRNA-seq里细胞类型在我们的scATAC-seq聚类中分布情况。这一步的目标是使用无约束整合的方法找到scATAC-seq和scRNA-seq中和T细胞和NK细胞对应的聚类,后续会用到该信息进行约束整合。具体操作为,我们创建一个confusionMatrix,并关注Cluster和predictedGroup_Un的交叉部分中scRNA-seq的细胞类型。
cM <- as.matrix(confusionMatrix(projHeme2$Clusters, projHeme2$predictedGroup_Un))
preClust <- colnames(cM)[apply(cM, 1 , which.max)]
cbind(preClust, rownames(cM)) #Assignments
输出信息如下,展示了12个scATAC-seq聚类中对应最优可能的scRNA-seq细胞类型。
# preClust
# [1,] "03_Late.Eryth" "C10"
# [2,] "20_CD4.N1" "C8"
# [3,] "16_Pre.B" "C3"
# [4,] "08_GMP.Neut" "C11"
# [5,] "17_B" "C4"
# [6,] "11_CD14.Mono.1" "C1"
# [7,] "01_HSC" "C12"
# [8,] "22_CD4.M" "C9"
# [9,] "09_pDC" "C5"
# [10,] "25_NK" "C7"
# [11,] "12_CD14.Mono.2" "C2"
# [12,] "06_CLP.1" "C6"
首先,我们检查在无约束整合中用到的scRNA-seq数据里细胞类型标签。
unique(unique(projHeme2$predictedGroup_Un))
# [1] "08_GMP.Neut" "25_NK" "16_Pre.B" "06_CLP.1"
# [5] "07_GMP" "11_CD14.Mono.1" "04_Early.Baso" "22_CD4.M"
# [9] "03_Late.Eryth" "05_CMP.LMPP" "17_B" "19_CD8.N"
#[13] "09_pDC" "13_CD16.Mono" "23_CD8.EM" "12_CD14.Mono.2"
#[17] "20_CD4.N1" "02_Early.Eryth" "21_CD4.N2" "24_CD8.CM"
#[21] "01_HSC"
从上面的输出中,我们发现scRNA-seq数据中与NK细胞和T细胞对应的聚类是Cluster 19 - 25。
接着我们创建一个字符串模式用来表示这些聚类,后续的约束整合会用到,其中|在正则表达式中表示"或",我们之后使用grep根据这些字符串模式从scATAC-seq提取和scRNA-seq对应的聚类。。
#From scRNA
cTNK <- paste0(paste0(19:25), collapse="|")
cTNK
# [1] "19|20|21|22|23|24|25"
其余的聚类就称之为"Non-T cell, Non-NK cell"(例如Cluster 1 - 18),也创建了对应的字符串模式
cNonTNK <- paste0(c(paste0("0", 1:9), 10:13, 15:18), collapse="|")
cNonTNK
#[1] "01|02|03|04|05|06|07|08|09|10|11|12|13|15|16|17|18"
接着再用字符串模式在preClust找到对应的scATAC-seq列名,然后使用列名从混合矩阵提取对应的列。
对于T细胞和NK细胞,scATAC-seq聚类ID就是C7, C8, C9
#Assign scATAC to these categories
clustTNK <- rownames(cM)[grep(cTNK, preClust)]
clustTNK
#[1] "C8" "C9" "C7"
对于" Non-T cells and Non-NK cells", ID就是scATAC-seq聚类余下的部分
clustNonTNK <- rownames(cM)[grep(cNonTNK, preClust)]
clustNonTNK
# [1] "C10" "C3" "C11" "C4" "C1" "C12" "C5" "C2" "C6"
接着在scRNA-seq中做相同的操作,筛选出相同的细胞类型。首先,我们鉴定scRNA-seq数据中T细胞和NK细胞
#RNA get cells in these categories
rnaTNK <- colnames(seRNA)[grep(cTNK, colData(seRNA)$BioClassification)]
head(rnaTNK)
#[1] "PBMC_10x_GREENLEAF_REP1:AAACCCAGTCGTCATA-1"
#[2] "PBMC_10x_GREENLEAF_REP1:AAACCCATCCGATGTA-1"
#[3] "PBMC_10x_GREENLEAF_REP1:AAACCCATCTCAACGA-1"
#[4] "PBMC_10x_GREENLEAF_REP1:AAACCCATCTCTCGAC-1"
#[5] "PBMC_10x_GREENLEAF_REP1:AAACGAACAATCGTCA-1"
#[6] "PBMC_10x_GREENLEAF_REP1:AAACGAACACGATTCA-1"
然后,鉴定scRNA-seq数据中"Non-T cell Non-NK cell cells"
rnaNonTNK <- colnames(seRNA)[grep(cNonTNK, colData(seRNA)$BioClassification)]
head(rnaNonTNK)
#[1] "CD34_32_R5:AAACCTGAGTATCGAA-1" "CD34_32_R5:AAACCTGAGTCGTTTG-1"
#[3] "CD34_32_R5:AAACCTGGTTCCACAA-1" "CD34_32_R5:AAACGGGAGCTTCGCG-1"
#[5] "CD34_32_R5:AAACGGGAGGGAGTAA-1" "CD34_32_R5:AAACGGGAGTTACGGG-1"
约束整合需要我们提供一个嵌套list。这是一个由多个SimpleList对象组成的SimpleList, 每一组对应一个约束情况。在案例中,我们有两个组,一个组称之为TNK,包括两个平台的T和NK细胞,另一个组为NonTNK,包括两个平台的"Non-T cell Non-NK cell"细胞。每个SimpleList对象都包含两个细胞ID的向量,一个是ATAC,一个是RNA.
groupList <- SimpleList(
TNK = SimpleList(
ATAC = projHeme2$cellNames[projHeme2$Clusters %in% clustTNK],
RNA = rnaTNK
),
NonTNK = SimpleList(
ATAC = projHeme2$cellNames[projHeme2$Clusters %in% clustNonTNK],
RNA = rnaNonTNK
)
)
我们将该列表传递给addGeneIntegrationMatrix()函数的groupList参数。注意,我们依旧没有将结果添加到Arrow文件中 (addToArrow = FALSE)。我们强烈建议,在保存到Arrow文件前先彻底的检查结果,看结果是否符合预期。在教程的下一节会介绍该操作。
projHeme2 <- addGeneIntegrationMatrix(
ArchRProj = projHeme2,
useMatrix = "GeneScoreMatrix",
matrixName = "GeneIntegrationMatrix",
reducedDims = "IterativeLSI",
seRNA = seRNA,
addToArrow = FALSE,
groupList = groupList,
groupRNA = "BioClassification",
nameCell = "predictedCell_Co",
nameGroup = "predictedGroup_Co",
nameScore = "predictedScore_Co"
)
8.1.3 约束整合和无约束整合对比
正如之前所提到的,我们的scATAC-seq数据可以根据整合的scRNA-seq数据进行定义,并且有约束和无约束这两种方式。为了对两者进行对比,我们分别根据这两种整合结果对scATAC-seq的数据进行上色。
首先,使用ArchR内置的paletteDiscrete()函数生成调色板
pal <- paletteDiscrete(values = colData(seRNA)$BioClassification)
在ArchR中,调色板本质上一个命名向量,每个十六进制编码的颜色对应着一个名字。
pal
# 01_HSC 02_Early.Eryth 03_Late.Eryth 04_Early.Baso 05_CMP.LMPP
# "#D51F26" "#502A59" "#235D55" "#3D6E57" "#8D2B8B"
# 06_CLP.1 07_GMP 08_GMP.Neut 09_pDC 10_cDC
# "#DE6C3E" "#F9B712" "#D8CE42" "#8E9ACD" "#B774B1"
#11_CD14.Mono.1 12_CD14.Mono.2 13_CD16.Mono 14_Unk 15_CLP.2
# "#D69FC8" "#C7C8DE" "#8FD3D4" "#89C86E" "#CC9672"
# 16_Pre.B 17_B 18_Plasma 19_CD8.N 20_CD4.N1
# "#CF7E96" "#A27AA4" "#CD4F32" "#6B977E" "#518AA3"
# 21_CD4.N2 22_CD4.M 23_CD8.EM 24_CD8.CM 25_NK
# "#5A5297" "#0F707D" "#5E2E32" "#A95A3C" "#B28D5C"
# 26_Unk
# "#3D3D3D"
我们在scATAC-seq数据根据无约束整合得到的scRNA-seq细胞类型进行可视化
p1 <- plotEmbedding(
projHeme2,
colorBy = "cellColData",
name = "predictedGroup_Un",
pal = pal
)
p1

do.call(cowplot::plot_grid, c(list(ncol = 3), p2c))
同样,我们也可以根据约束整合得到scATAC-seq对应的scRNA-seq的细胞类型进行可视化
p2 <- plotEmbedding(
projHeme2,
colorBy = "cellColData",
name = "predictedGroup_Co",
pal = pal
)
p2

这两者的结果差异其实非常细微,主要是我们感兴趣的细胞类型原本就存在明显的差异。当然,仔细观察还能发现其中的不同之处,尤其是T细胞(Clusters 17-22)
我们用plotPDF()函数保存该图矢量版本。
plotPDF(p1,p2, name = "Plot-UMAP-RNA-Integration.pdf", ArchRProj = projHeme2, addDOC = FALSE, width = 5, height = 5)
我们现在可以用saveArchRProject()函数保存我们的projHeme2对象。
saveArchRProject(ArchRProj = projHeme2, outputDirectory = "Save-ProjHeme2", load = FALSE)
8.2 为每个scATAC-seq细胞增加拟scRNA-seq谱
既然我们对scATAC-seq和scRNA-seq整合的结果感到满意,我们就能用addToArrow=TRUE重新运行,在Arrow文件中添加相关联的表达量矩阵数据。根据之前所提到的,我们传入groupList约束整合,在nameCell,nameGroup和nameScore中加入列名。这些元信息列都会被添加到cellColData中。
这里,我们新建了一个projHeme3,用于后续教程。
#~5 minutes
projHeme3 <- addGeneIntegrationMatrix(
ArchRProj = projHeme2,
useMatrix = "GeneScoreMatrix",
matrixName = "GeneIntegrationMatrix",
reducedDims = "IterativeLSI",
seRNA = seRNA,
addToArrow = TRUE,
force= TRUE,
groupList = groupList,
groupRNA = "BioClassification",
nameCell = "predictedCell",
nameGroup = "predictedGroup",
nameScore = "predictedScore"
)
现在,当我们使用getAvailableMatrices()检查哪些矩阵可用时,我们会发现GeneIntegrationMatrix已经被添加到Arrow文件中
getAvailableMatrices(projHeme3)
# [1] "GeneIntegrationMatrix" "GeneScoreMatrix" "TileMatrix"
在新的GeneIntegrationMatrix中,我们可以比较连接的基因表达量和根据基因得分推断的基因表达量
我们需要先确保在项目中加入了填充权重值(impute weights)
projHeme3 <- addImputeWeights(projHeme3)
现在,我们来生成一些UMAP图,里面的基因表达量值来自于GeneIntegrationMatrix
markerGenes <- c(
"CD34", #Early Progenitor
"GATA1", #Erythroid
"PAX5", "MS4A1", #B-Cell Trajectory
"CD14", #Monocytes
"CD3D", "CD8A", "TBX21", "IL7R" #TCells
)
p1 <- plotEmbedding(
ArchRProj = projHeme3,
colorBy = "GeneIntegrationMatrix",
name = markerGenes,
continuousSet = "horizonExtra",
embedding = "UMAP",
imputeWeights = getImputeWeights(projHeme3)
)
以及一些相同UMAP图,但是使用GeneScoreMatrix里的基因得分值
p2 <- plotEmbedding(
ArchRProj = projHeme3,
colorBy = "GeneScoreMatrix",
continuousSet = "horizonExtra",
name = markerGenes,
embedding = "UMAP",
imputeWeights = getImputeWeights(projHeme3)
)
最后用cowplot将这些标记基因绘制在一起
p1c <- lapply(p1, function(x){
x + guides(color = FALSE, fill = FALSE) +
theme_ArchR(baseSize = 6.5) +
theme(plot.margin = unit(c(0, 0, 0, 0), "cm")) +
theme(
axis.text.x=element_blank(),
axis.ticks.x=element_blank(),
axis.text.y=element_blank(),
axis.ticks.y=element_blank()
)
})
p2c <- lapply(p2, function(x){
x + guides(color = FALSE, fill = FALSE) +
theme_ArchR(baseSize = 6.5) +
theme(plot.margin = unit(c(0, 0, 0, 0), "cm")) +
theme(
axis.text.x=element_blank(),
axis.ticks.x=element_blank(),
axis.text.y=element_blank(),
axis.ticks.y=element_blank()
)
})
do.call(cowplot::plot_grid, c(list(ncol = 3), p1c))

do.call(cowplot::plot_grid, c(list(ncol = 3), p2c))

和预期的一样,两个方法推测的基因表达量存在相似性,但并不相同。
使用plotPDF()函数保存可编辑的矢量版。
plotPDF(plotList = p1,
name = "Plot-UMAP-Marker-Genes-RNA-W-Imputation.pdf",
ArchRProj = projHeme3,
addDOC = FALSE, width = 5, height = 5)
8.3 使用scRNA-seq信息标记scATAC-seq聚类
现在,我们确定了scATAC-seq和scRNA-seq数据间的对应关系,我们就可以使用scRNA-seq数据中细胞类型对我们的scATAC-seq聚类进行定义。
首先,我们会在scATAC-seq和整合分析得到predictedGroup之间构建一个混合矩阵
cM <- confusionMatrix(projHeme3$Clusters, projHeme3$predictedGroup)
labelOld <- rownames(cM)
labelOld
# [1] "Cluster11" "Cluster2" "Cluster12" "Cluster1" "Cluster8" "Cluster4"
# [7] "Cluster9" "Cluster5" "Cluster7" "Cluster14" "Cluster3" "Cluster10"
# [13] "Cluster6" "Cluster13"
接着,对于每一个scATAC-seq聚类,我们根据predictedGroup确定最能定义聚类的细胞类型。
labelNew <- colnames(cM)[apply(cM, 1, which.max)]
labelNew
接着,我们需要对新的聚类标签进行重命名,简化分类系统。对于每一个scRNA-seq的聚类,我们会重新定义标签,以便更好解释。
remapClust <- c(
"01_HSC" = "Progenitor",
"02_Early.Eryth" = "Erythroid",
"03_Late.Eryth" = "Erythroid",
"04_Early.Baso" = "Basophil",
"05_CMP.LMPP" = "Progenitor",
"06_CLP.1" = "CLP",
"07_GMP" = "GMP",
"08_GMP.Neut" = "GMP",
"09_pDC" = "pDC",
"10_cDC" = "cDC",
"11_CD14.Mono.1" = "Mono",
"12_CD14.Mono.2" = "Mono",
"13_CD16.Mono" = "Mono",
"15_CLP.2" = "CLP",
"16_Pre.B" = "PreB",
"17_B" = "B",
"18_Plasma" = "Plasma",
"19_CD8.N" = "CD8.N",
"20_CD4.N1" = "CD4.N",
"21_CD4.N2" = "CD4.N",
"22_CD4.M" = "CD4.M",
"23_CD8.EM" = "CD8.EM",
"24_CD8.CM" = "CD8.CM",
"25_NK" = "NK"
)
remapClust <- remapClust[names(remapClust) %in% labelNew]
接着,使用mapLables()函数进行标签转换,将旧的标签映射到新的标签上。
labelNew2 <- mapLabels(labelNew, oldLabels = names(remapClust), newLabels = remapClust)
labelNew2
# [1] "GMP" "B" "PreB" "CD4.N" "Mono"
# [6] "Erythroid" "Progenitor" "CD4.M" "pDC" "NK"
# [11] "CLP" "Mono"
合并labelsOld和labelsNew2,我们现在可以用mapLables()函数在cellColData里新建聚类标签。
projHeme3$Clusters2 <- mapLabels(projHeme3$Clusters, newLabels = labelNew2, oldLabels = labelOld)
得到新的标签后,我们绘制最新的UMAP展示聚类结果。
p1 <- plotEmbedding(projHeme3, colorBy = "cellColData", name = "Clusters2")
p1
如果被分析scATAC-seq数据对应的细胞系统也有scRNA-seq数据存在,那么这种分析范式将会特别有用。正如之前所说,这种scRNA-seq和scATAC-seq整合分析也为后续更加复杂的基因调控分析提供了出色的框架。
用plotPDF()函数保存该图矢量版本。
plotPDF(p1, name = "Plot-UMAP-Remap-Clusters.pdf", ArchRProj = projHeme2, addDOC = FALSE, width = 5, height = 5)
使用saveArchRProject保存我们最初的projHeme3.
saveArchRProject(ArchRProj = projHeme3, outputDirectory = "Save-ProjHeme3", load = FALSE)
