多倍化以及后续的基因丢失和二倍化现象存在于大部分的物种中, 是物种进化的重要动力。如果一个物种在演化过程中发生过多倍化,那么在基因组上就会存在一些共线性区域(即两个区域之间的基因是旁系同源基因,其基因的排布顺序基本一致)。
例如拟南芥经历了3次古多倍化,包括2次二倍化,一次3倍化 (Tang. et.al 2008 Science).
..For example, Arabidopsis thaliana (thale cress) has undergone three paleo-polyploidies, including two doublings (5) and one tripling (12), resulting in ~12 copies of its ancestral chromosome set in a ~160-Mb genome...
当然之前只是记住了结论,现在我想的是,如何复现出这个分析结果呢?
前期准备
首先,我们需要使用conda安装wgdi。 我一般会新建一个环境避免新装软件和其他软件产生冲突
# 安装软件
conda create -c bioconda -c conda-forge -n wgdi wgdi
# 启动环境
conda activate wgdi
之后,我们从ENSEMBL上下载基因组,CDS, 蛋白和GFF文件
#Athaliana
wget ftp://ftp.ensemblgenomes.org/pub/plants/release-44/fasta/arabidopsis_thaliana/dna/Arabidopsis_thaliana.TAIR10.dna.toplevel.fa.gz
gunzip Arabidopsis_thaliana.TAIR10.dna.toplevel.fa.gz
wget ftp://ftp.ensemblgenomes.org/pub/plants/release-44/fasta/arabidopsis_thaliana/cds/Arabidopsis_thaliana.TAIR10.cds.all.fa.gz
gunzip Arabidopsis_thaliana.TAIR10.cds.all.fa.gz
wget ftp://ftp.ensemblgenomes.org/pub/plants/release-44/fasta/arabidopsis_thaliana/pep/Arabidopsis_thaliana.TAIR10.pep.all.fa.gz
gunzip Arabidopsis_thaliana.TAIR10.pep.all.fa.gz
wget ftp://ftp.ensemblgenomes.org/pub/plants/release-44/gff3/arabidopsis_thaliana/Arabidopsis_thaliana.TAIR10.44.gff3.gz
gunzip Arabidopsis_thaliana.TAIR10.44.gff3.gz
注意: cDNA和CDS并不相同,CDS只包括起始密码子到终止密码子之间的序列,而cDNA还会包括UTR.
数据预处理
数据的预处理是用时最长的步骤,因为软件要求的输入格式并非是你手头所拥有的格式,你通常都需要进行一些转换才能得到它所需的形式。
wgdi需要三种信息,分别是BLAST, 基因的位置信息和染色体长度信息,要求格式如下
-
BLAST 的
-outfmt 6输出的文件 -
基因的位置信息: 以tab分隔,分别为chr,id,start,end,strand,order,old_id。(并非真正意义上的GFF格式)
-
染色体长度信息和染色体上的基因个数,格式为 chr, length, gene number
同时,对于每个基因我们只需要一个转录本,我通常使用最长的转录本作为该基因的代表。之前我写过一个脚本 (get_the_longest_transcripts.py) 提取每个基因的最长转录本,我在此处上写了一个新的脚本用来根据参考基因组和注释的GFF文件生成wgdi的两个输入文件,脚本地址为https://github.com/xuzhougeng/myscripts/blob/master/comparative/generate_conf.py
pytho generate_conf.py -p ath Arabidopsis_thaliana.TAIR10.dna.toplevel.fa Arabidopsis_thaliana.TAIR10.44.gff3
输出文件是ath.gff 和 ath.len.
由于ENSEMBL上GFF的ID编号和pep.fa 和cds.fa 不太一致,简单的说就是,编号之前中有一个 "gene:" 和 "transcript:" . 如下
$ head ath.gff
1 gene:AT1G01010 3631 5899 + 1 transcript:AT1G01010.1
1 gene:AT1G01020 6788 9130 - 2 transcript:AT1G01020.1
1 gene:AT1G01030 11649 13714 - 3 transcript:AT1G01030.1
1 gene:AT1G01040 23416 31120 + 4 transcript:AT1G01040.2
1 gene:AT1G01050 31170 33171 - 5 transcript:AT1G01050.1
1 gene:AT1G01060 33379 37757 - 6 transcript:AT1G01060.3
1 gene:AT1G01070 38444 41017 - 7 transcript:AT1G01070.1
1 gene:AT1G01080 45296 47019 - 8 transcript:AT1G01080.2
1 gene:AT1G01090 47234 49304 - 9 transcript:AT1G01090.1
1 gene:AT1G01100 49909 51210 - 10 transcript:AT1G01100.2
于是我用 sed 删除了这些信息
sed -i -e 's/gene://' -e 's/transcript://' ath.gff
另外,pep.fa, cds.fa里面包含所有的基因,且命名特别的长, 同时我们也不需要基因中的 ".1", ".2" 这部分信息
$ head -n 5 Arabidopsis_thaliana.TAIR10.cds.all.fa
>AT3G05780.1 cds chromosome:TAIR10:3:1714941:1719608:-1 gene:AT3G05780 gene_biotype:protein_coding transcript_biotype:protein_coding gene_symbol:LON3 description:Lon protease homolog 3, mitochondrial [Source:UniProtKB/Swiss-Prot;Acc:Q9M9L8]
ATGATGCCTAAACGGTTTAACACGAGTGGCTTTGACACGACTCTTCGTCTACCTTCGTAC
TACGGTTTCTTGCATCTTACACAAAGCTTAACCCTAAATTCCCGCGTTTTCTACGGTGCC
CGCCATGTGACTCCTCCGGCTATTCGGATCGGATCCAATCCGGTTCAGAGTCTACTACTC
TTCAGGGCACCGACTCAGCTTACCGGATGGAACCGGAGTTCTCGCGATTTATTGGGTCGTb
借助于seqkit, 我对原来的数据进行了筛选和重命名
seqkit grep -f <(cut -f 7 ath.gff ) Arabidopsis_thaliana.TAIR10.cds.all.fa | seqkit seq --id-regexp "^(.*?)\\.\\d" -i > ath.cds.fa
seqkit grep -f <(cut -f 7 ath.gff ) Arabidopsis_thaliana.TAIR10.pep.all.fa | seqkit seq --id-regexp "^(.*?)\\.\\d" -i > ath.pep.fa
通过NCBI的BLASTP 或者DIAMOND 进行蛋白之间相互比对,输出格式为 -outfmt 6
makeblastdb -in ath.pep.fa -dbtype prot
blastp -num_threads 50 -db ath.pep.fa -query ath.pep.fa -outfmt 6 -evalue 1e-5 -num_alignments 20 -out ath.blastp.txt &
共线性分析
绘制点阵图
在上面步骤结束后,其实绘制点阵图就非常简单了,只需要创建配置文件,然后修改配置文件,最后运行wgdi。
基本wgdi的其他分析也都是三部曲,创建配置,修改配置,运行程序。
第一步,创建配置文件
wgdi -d \? > ath.conf
配置文件的信息如下,
[dotplot]
blast = blast file
gff1 = gff1 file
gff2 = gff2 file
lens1 = lens1 file
lens2 = lens2 file
genome1_name = Genome1 name
genome2_name = Genome2 name
multiple = 1 # 最好的同源基因数, 用输出结果中会用红点表示
score = 100 # blast输出的score 过滤
evalue = 1e-5 # blast输出的evalue 过滤
repeat_number = 20 # genome2相对于genome1的最多同源基因数
position = order
blast_reverse = false
ancestor_left = none
ancestor_top = none
markersize = 0.5 # 点的大小
figsize = 10,10 # 图片大小
savefig = savefile(.png,.pdf)
第二步,修改配置文件。因为是自我比对,所以 gff1和gff2内容一样, lens1和lens2内容一样
[dotplot]
blast = ath.blastp.txt
gff1 = ath.gff
gff2 = ath.gff
lens1 = ath.len
lens2 = ath.len
genome1_name = A. thaliana
genome2_name = A. thaliana
multiple = 1
score = 100
evalue = 1e-5
repeat_number = 5
position = order
blast_reverse = false
ancestor_left = none
ancestor_top = none
markersize = 0.5
figsize = 10,10
savefig = ath.dot.png
最后运行如下命令
wgdi -d ath.conf
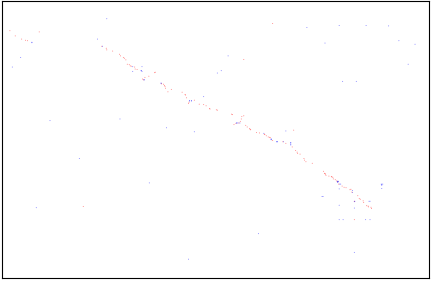
输出的png如下所示

从图中,我们很容易地就能观察到三种颜色,分别是红色,蓝色,和灰色。在WGDI中,红色表示genome2的基因在genome1中的最优同源(相似度最高)的匹配,次好的四个基因是蓝色,而剩余部分是为灰色。图中对角线出现的片段并非是自身比对,因为WGDI已经过滤掉自身比自身的结果。那么问题来了,这些基因是什么呢?这个问题会在后续的分析中进行解答。
如果基因组存在加倍事件,那么对于一个基因组的某个区域可能会存在另一个区域与其有着相似的基因(同源基因),并且这些基因的排布顺序较为一致。反应到点阵图上,就是能够观察到有多个点所组成的"线"。这里的线需要有一个引号,因为当你放大之后,你会发现其实还是点而已。

由于进一步鉴定共线性区域就建立在这些"点"的基础上,那么影响这些点是否为同源基因的参数就非常重要了,即配置文件中的score,evalue,repeat_number。
共线性分析和Ks计算
对于同线性(synteny)和共线性(collinearity),一般认为同线性指的是两个区域有一定数量的同源基因,对基因顺序排布无要求。共线性是同线性的特殊形式,要求同源基因的排布顺序也相似。
wgdi开发了-icl模块(Improved version of ColinearScan)用于进行共线性分析,使用起来非常简单,也是先建立配置文件。
wgdi -icl \? >> ath.conf
然后对配置文件进行修改.
[collinearity]
gff1 = ath.gff
gff2 = ath.gff
lens1 = ath.len
lens2 = ath.len
blast = ath.blastp.txt
blast_reverse = false
multiple = 1
process = 8
evalue = 1e-5
score = 100
grading = 50,40,25
mg = 40,40
pvalue = 0.2
repeat_number = 10
positon = order
savefile = ath.collinearity.txt
其中的evalue, score和BLAST文件的过滤有关,用于筛选同源基因对。multiple确定最佳比对基因,repeat_number表示最多允许多少基因是潜在的同源基因, grading根据同源基因的匹配程度进行打分, mg 指的是共线性区域中所允许最大空缺基因数。
运行结果后,会得到ath.collinearity.txt,记录共线性区域。以 # 起始的行记录共线性区域的元信息,例如得分(score),显著性(pvalue),基因对数(N)等。
grep '^#' ath.collinearity.txt | tail -n 1
# Alignment 959: score=778.0 pvalue=0.0003 N=16 Pt&Pt minus
紧接着,我们就可以根据共线性结果来计算Ks。
同样的也是先生成配置文件
wgdi -ks \? >> ath.conf
然后修改配置文件。我们需要提供cds和pep文件,共线性分析输出文件(支持MCScanX的共线性分析结果)。WGDI会用muscle根据蛋白序列进行联配,然后使用pal2pal.pl 基于cds序列将蛋白联配转成密码子联配,最后用paml中的yn00计算ka和ks。
[ks]
cds_file = ath.cds.fa
pep_file = ath.pep.fa
align_software = muscle
pairs_file = ath.collinearity.txt
ks_file = ath.ks
ks计算需要一段时间,那么不妨在计算时了解下Ka和Ks。Ka和Ks分别指的是非同义替换位点数,和同义替换位点数。根据中性演化假说,基因的变化大多是中性突变,不会影响生物的生存,那么当一对同源基因分开的时间越早,不影响生存的碱基替换数就越多(Ks),反之越多。
运行结果后,输出的Ks文件共有6列,对应的是每个基因对的Ka和Ks值。
$ head ath.ks
id1 id2 ka_NG86 ks_NG86 ka_YN00 ks_YN00
AT1G72300 AT1G17240 0.1404 0.629 0.1467 0.5718
AT1G72330 AT1G17290 0.0803 0.5442 0.079 0.6386
AT1G72350 AT1G17310 0.3709 0.9228 0.3745 1.071
AT1G72410 AT1G17360 0.2636 0.875 0.2634 1.1732
AT1G72450 AT1G17380 0.2828 1.1068 0.2857 1.5231
AT1G72490 AT1G17400 0.255 1.2113 0.2597 2.0862
AT1G72520 AT1G17420 0.1006 0.9734 0.1025 1.0599
AT1G72620 AT1G17430 0.1284 0.7328 0.1375 0.603
AT1G72630 AT1G17455 0.0643 0.7608 0.0613 1.2295
鉴于共线性和Ks值输出结果信息量太大,不方便使用,更好的方法是将两者进行聚合,得到关于各个共线性区的汇总信息。
WGDI提供 -bi 参数帮助我们进行数据整合。同样也是生成配置文件
wgdi -bi ? >> ath.conf
修改配置文件. 其中collinearity 和ks是我们前两步的输出文件,而 ks_col 则是声明使用ks文件里的哪一列。
[blockinfo]
blast = ath.blastp.txt
gff1 = ath.gff
gff2 = ath.gff
lens1 = ath.len
lens2 = ath.len
collinearity = ath.collinearity.txt
score = 100
evalue = 1e-5
repeat_number = 20
position = order
ks = ath.ks
ks_col = ks_NG86
savefile = ath_block_information.csv
运行即可
wgdi -bi ath.conf
输出文件以csv格式进行存放,因此可以用EXCEL直接打开,共11列。
id即共线性的结果的唯一标识chr1,start1,end1即参考基因组(点图的左边)的共线性范围chr2,start2,end2即参考基因组(点图的上边)的共线性范围pvalue即共线性结果评估,常常认为小于0.01的更合理些length即共线性片段长度ks_median即共线性片段上所有基因对ks的中位数(主要用来评判ks分布的)ks_average即共线性片段上所有基因对ks的平均值homo1,homo2,homo3,homo4,homo5与multiple参数有关,即一共有homo+multiple列
主要规则是:基因对如果在点图中为红色,赋值为1,蓝色赋值为0,灰色赋值为-1(颜色参考前述 wgdi 点图 相关推文)。共线性片段上所有基因对赋值后求平均值,这样就赋予该共线性一个-1,1的值。如果共线性的点大部分为红色,那么该值接近于1。可以作为共线性片段的筛选。
block1,block2分别为共线性片段上基因order的位置。ks共线性片段的上基因对的ks值density1,density2共线性片段的基因分布密集程度。值越小表示稀疏
这个表格是后续探索分析的一个重点,比如说我们可以用它来分析我们点图中出现在对角线上的基因。下面的代码就是提取了第一个共线性区域的所有基因,然后进行富集分析,绘制了一个气泡图。
df <- read.csv("ath_block_information.csv")
tandem_length <- 200
df$start <- df$start2 - df$start1
df$end <- df$end2 - df$end1
df_tandem <- df[abs(df$start) <= tandem_length |
abs(df$end) <= tandem_length,]
gff <- read.table("ath.gff")
syn_block1 <- df_tandem[1,c("block1", "block2")]
gene_order <- unlist(
lapply(syn_block1, strsplit, split="_", fixed=TRUE, useBytes=TRUE)
)
gene_order <- unique(as.numeric(gene_order))
syn_block1_gene <- gff[ gff$V6 %in% gene_order, "V2"]
#BiocManager::install("org.At.tair.db")
library(org.At.tair.db)
library(clusterProfiler)
ego <- enrichGO(syn_block1_gene,
OrgDb = org.At.tair.db,
keyType = "TAIR",ont = "BP"
)
dotplot(ego)

进一步观察这些基因,可以发现这些基因的编号都是前后相连,说明这个基因簇和花粉识别有一定关系。
> as.data.frame(ego)[1,]
ID Description GeneRatio BgRatio pvalue
GO:0048544 GO:0048544 recognition of pollen 11/560 43/21845 7.794138e-09
p.adjust qvalue
GO:0048544 8.121073e-06 7.95418e-06
geneID
GO:0048544 AT1G61360/AT1G61370/AT1G61380/AT1G61390/AT1G61400/AT1G61420/AT1G61440/AT1G61480/AT1G61490/AT1G61500/AT1G61550
Count
GO:0048544 11
A gene cluster is a group of two or more genes found within an organism's DNA that encode similar polypeptides, or proteins, which collectively share a generalized function and are **often located within a few thousand base pairs **of each other. --维基百科
接下来,我们还可以根据这个表格中的Ks对点阵图进行上色, 以及正确的计算Ks峰值。
