利用3D-DNA流程组装基因组
使用二代数据或三代数据得到contig后,下一步就是将contig提升到染色体水平。有很多策略可以做到这一点,比如说遗传图谱,BioNano(看运气), HiC, 参考近源物种。
如果利用HiC进行准染色体水平,那么目前常见的组装软件有下面几个
- HiRise: 2015年后的GitHub就不再更新
- LACHESIS: 发表在NBT,2017年后不再更新
- SALSA: 发表在BMC genomics, 仍在更新中
- 3D-DNA: 发表在science,仍在更新中
- ALLHiC: 发表在Nature Plants, 用于解决植物多倍体组装问题
对于二倍体物种而言,目前3D-DNA应该是组装效果最好的一个软件。
工作流程
使用3D-DNA做基因组组装的整体流程如下图,分别为组装,Juicer分析Hi-C数据,3D-DNA进行scaffolding,使用JBAT对组装结果进行手工纠正,最终得到准染色体水平的基因组。
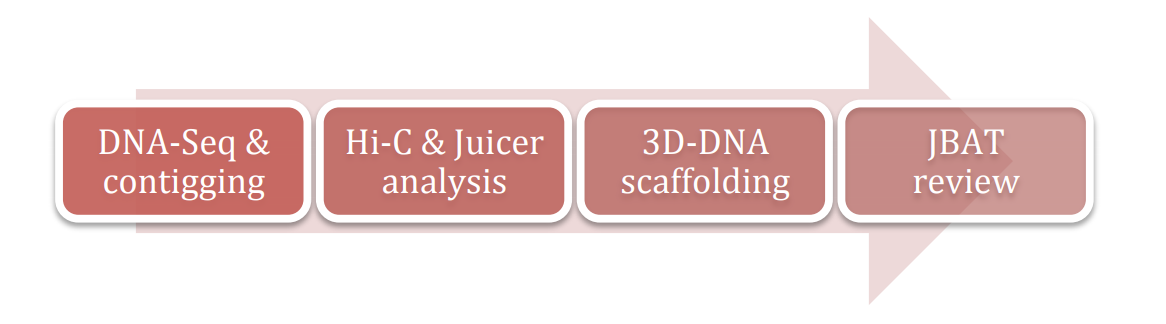
基因组组装可以是二代测序方法,也可以是三代测序组装方法,总之会得到contig。
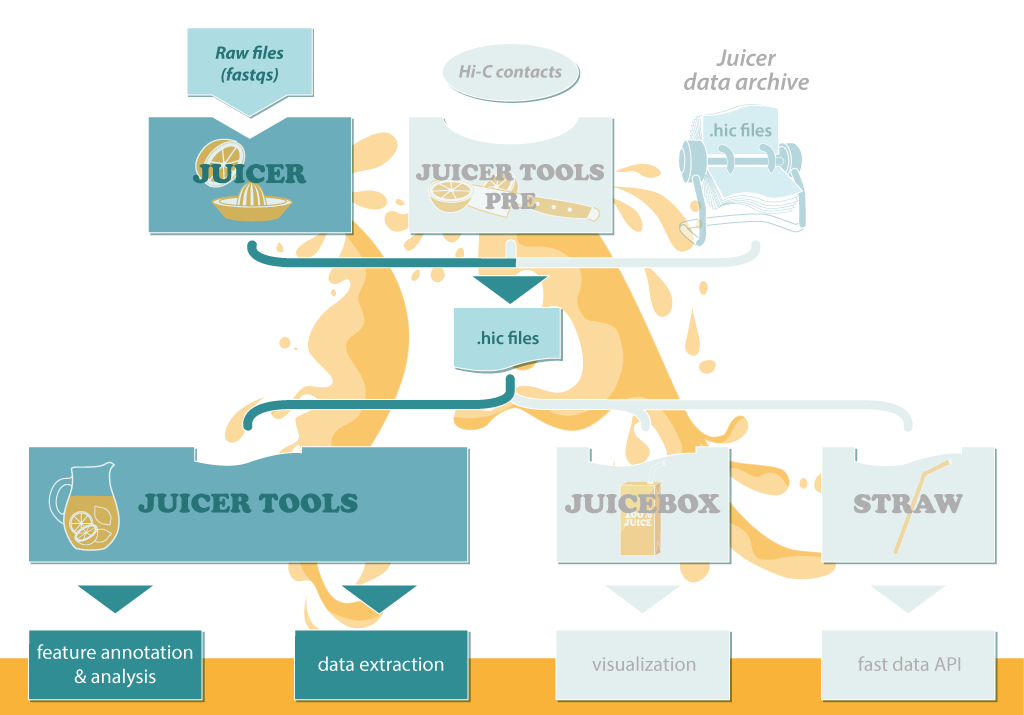
Juicer的工作流程见下图,输入原始的fastq文件,处理得到中间文件.hic, 之后对.hic文件用于下游分析,包括
- Arrowhead: 寻找存在关联的区域
- HiCCUPS: 分析局部富集peaks
- MotifFinder: 用于锚定peaks
- Persons: 计算观测/期望的皮尔森相关系数矩阵
- Eigenvector: 确定分隔
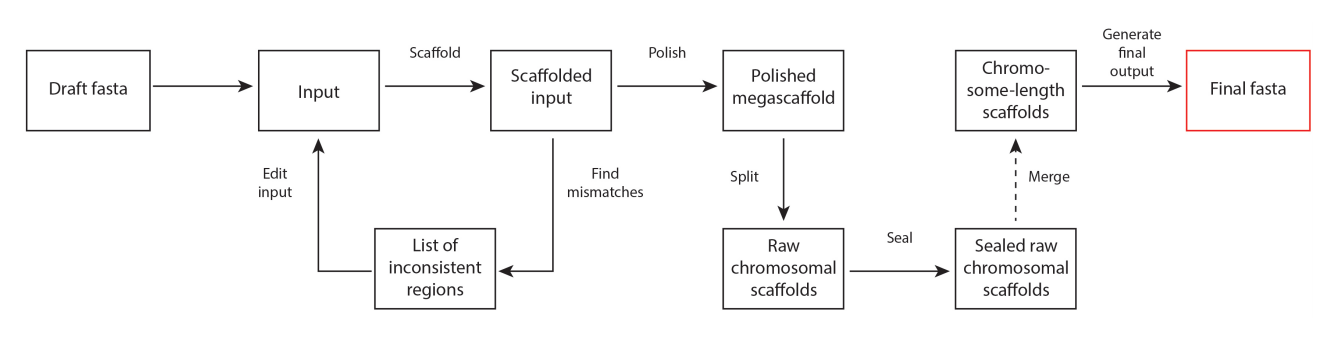
之后Juicer的输出结果给3D-DNA,分析流程见下图。3D-DNA先根据Hi-C数据分析contig中的misjoin,对其进行纠错。之后通过四步,分别是Polish, Split, Seal和Merge, 得到最终的基因组序列
软件安装
在安装之前,确保服务器上有了下面这些依赖软件工具
- LastZ(仅在杂合基因组的二倍体模式下使用)
- Java >= 1.7
- GNU Awk >= 4.02
- GNU coreutils sort > 8.11
- Python >= 2.7
- scipy, numpy, matplotlib
- GNU Parallel >=20150322 (不必要,但是强力推荐)
- bwa
我们需要安装两个软件,一个是3D-DNA,另一个是juicer。
CPU版本的juicer安装
mkdir -p ~/opt/biosoft/
cd ~/opt/biosoft
git clone https://github.com/theaidenlab/juicer.git
cd juicer
ln -s CPU scripts
cd scripts/common
wget https://hicfiles.tc4ga.com/public/juicer/juicer_tools.1.9.9_jcuda.0.8.jar
ln -s juicer_tools.1.9.9_jcuda.0.8.jar juicer_tools.jar
然后用~/opt/biosoft/juicer/scripts/juicer.sh -h检查是否有帮助信息输出
3D-DNA安装也很容易,只需要从Github上将内容克隆到本地即可
cd ~/opt/biosoft
git clone https://github.com/theaidenlab/3d-dna.git
用sh ~/opt/biosoft/3d-dna/run-asm-pipeline.sh -h查看是否有帮助文档输出。
参数详解
以CPU版本的为例,juicer.sh的参数如下
Usage: juicer.sh [-g genomeID] [-d topDir] [-s site] [-a about] [-R end]
[-S stage] [-p chrom.sizes path] [-y restriction site file]
[-z reference genome file] [-D Juicer scripts directory]
[-b ligation] [-t threads] [-r] [-h] [-f] [-j]
参数说明
-g: 定义一个物种名-s: 酶切类型, HindIII(AAGCTAGCTT), MboI(GATCGATC) , DpnII(GATCGATC), NcoI(CCATGCATGG)-z: 参考基因组文件-y: 限制性酶切位点可能出现位置文件-p: 染色体大小文件-C: 将原来的文件进行拆分,必须是4的倍数,默认是90000000, 即22.5M reads-S: 和任务重运行有关,从中途的某一步开始,“merge”, “dedup”, “final”, “postproc” 或 “early”-D: juicer的目录,我们安装在~/opt/biosoft/,所以设置为~/opt/biosoft/juicer-a: 实验的描述说明,可以不用设置-t: 线程数
juicer.sh还有AWS, LSF, PBS, SLURM版本,由于我的服务器是单主机,无法进行测试讲解。
如果你的基因组不是复杂基因组,比如说高杂合,高重复序列,或者Hi-C数据测太少,那么3d-dna的流程更加简单, run-asm-pipeline.sh -h只有四个参数需要改
-i|--input: 过滤长度低于给定阈值的contig/scaffold, 默认是15000-r|--round: 基因组中misjoin的纠错轮数,默认是2,当基因组比较准确时,设置为0,然后在JABT中调整会更好-m|--mode: 是否调用merge模块,当且仅当在杂合度比较高的情况下使用,也就是组装的单倍型基因组明显偏大-s|--stage: 从polish, split, seal, merge 或finalize 的某一个阶段开始
但是,一旦基因组复杂起来,那么需要调整的参数就非常多了, run-asm-pipeline.sh --help会输出更多的信息,你需要根据当前结果去确定每个阶段的参数应该如何调整。
最终的输出文件最关键的是下面三类:
.fasta: 以FINAL标记的是最终结果.hic: 各个阶段都会有输出结果,用于在JABT中展示.assembly: 各个阶段都会有输出,一共两列,存放contig的组装顺序
分析过程
假如你现在目录下有2个文件夹,reference
- reference: 存放一个genome.fa, 为组装的contigs
- fastq: 存放HiC二代双端测序结果,read_R1_fastq.gz, read_R2_fastq.gz
注意,genome.fa中的序列一定得是80个字符分隔的情况,也就是多行FASTA。
增加一个新的基因组
第一步: 为基因组建立BWA索引
cd reference
bwa index genome.fa
第二步: 根据基因组构建创建可能的酶切位点文件
python ~/opt/biosoft/juicer/misc/generate_site_positions.py DpnII genome genome.fa
第三步: 运行如下命令, 获取每条contig的长度
awk 'BEGIN{OFS="\t"}{print $1, $NF}' genome_DpnII.txt > genome.chrom.size
# 返回上级目录
cd ..
运行juicer
保证当前目录下有fastq和reference文件夹,然后运行如下命令,一定要设置-z,-p,-y这三个参数
~/opt/biosoft/juicer/scripts/juicer.sh \
-g genome \
-s MboI \
-z reference/genome.fa \
-y reference/genome_DpnII.txt \
-p reference/genome.chrom.size \
-D ~/opt/biosoft/juicer \
-t 40 &> juicer.log &
你可能会好奇为啥这里出现两个酶,DpnII和MboI。这是因为DpnI, DpnII, MboI, Sau3AI, 识别相同的序列,GATC,仅仅是对甲基化敏感度不同。
输出的结果文件都在aligned目录下,其中"merged_nodups.txt"就是下一步3D-DNA的输入文件之一
运行3d-dna
3d-dna的运行也没有多少参数可以调整,如果对组装的信心高,就用-r 0, 否则用默认的-r 2就行了。
~/opt/biosoft/3d-dna/run-asm-pipeline.sh -r 2 reference/genome.fa aligned/merged_nodups.txt &> 3d.log &
然后在Juicer-Tools中对结果进行可视化,对可能的错误进行纠正
最后输出文件中,包含FINAL就是我们需要的结果。
使用juicerbox进行手工纠错
关于juicerbox的用法,我已经将原视频搬运到哔哩哔哩, 见https://www.bilibili.com/video/av65134634
最常见的几种组装错误:
- misjoin: 切割
- translocations: 移动
- inversions: 翻转
- chromosome boundaries: 确定染色体的边界
这些错误的判断依赖于经验,所以只能靠自己多试试了。
最后输出genome.review.assembly用于下一步的分析
再次运行3d-dna
根据JABT手工纠正的结果, genome.review.assembly, 使用run-asm-pipeline-post-review.sh重新组装基因组。
~/opt/biosoft/3d-dna/run-asm-pipeline-post-review.sh \
-r genome.review.assembly genome.fa aligned/merged_nodups.txt &> 3d.log &
个人使用评价
juicer的代码个人感觉不是特别的好,至少以下几个地方都需要改,
- 临时文件不会去及时删除
- bwa得到的SAM文件处理方式有待优化,使用BAM能更快的并行计算
- 参数命令的判断很差,用-z判断字符串是否为0,而不是用-f或-d去判断文件是否存在,这个我已经提了一个issue,希望能改吧
- Linux的sort支持多线程,但是没看到用
- 脚本中有些限速步骤的awk代码,不知道什么时候能改成更高效的处理
前两条导致了运行过程中要占用大量的硬盘,所以不准备2T左右的硬盘,很容易出错。第三条是一些报错不会及时停止运算,也不容易排查。估计公司从效率角度出发,应该是写了很多脚本来替换原来的awk脚本了
另外,juicer在多倍体物种上表现很差,建议使用ALLHiC
参考资料
- https://github.com/theaidenlab/3d-dna
- https://github.com/aidenlab/juicer
- http://aidenlab.org/assembly/manual_180322.pdf
- https://www.neb.com/faqs/0001/01/01/what-s-the-difference-between-dpni-dpnii-mboi-and-sau3ai
假如你不小心设置了错误的-p参数,也不是特别的要紧,因为之后在最后阶段(final) 才会遇到了下面这个报错
Could not find chromosome sizes file for: reference/genome.chrom.size
***! Can't find inter.hic in aligned/inter_30.hic
***! Error! Either inter.hic or inter_30.hic were not created
Either inter.hic or inter_30.hic were not created. Check aligned for results
即便遇到了这个报错也不要紧,因为inter.hic 和 inter_30.hic在3d-dna流程中用不到,所以不需要解决。
如果需要解决的话,有两个解决方案,一种重新运行命令,只不过多加一个参数-S final, 就会跳过之前的比对,合并和去重步骤,直接到后面STATISTICS环节。但是这样依旧会有一些不必要的计算工作,所以另一种方法就是运行原脚本必要的代码
juiceDir=~/opt/biosoft/juicer
outputdir=aligned
genomePath=reference/genome.chrom.size
site_file=reference/genome_DpnII.txt
ligation=GATCGATC
# output is inter.hic
${juiceDir}/scripts/common/juicer_tools pre -f $site_file -s $outputdir/inter.txt -g $outputdir/inter_hists.m -q 1 $outputdir/merged_nodups.txt $outputdir/inter.hic $genomePath
# output is inter_30.txt
${juiceDir}/scripts/common/statistics.pl -s $site_file -l $ligation -o $outputdir/inter_30.txt -q 30 $outputdir/merged_nodups.txt
# output is inter_30.hic
${juiceDir}/scripts/common/juicer_tools pre -f $site_file -s $outputdir/inter_30.txt -g $outputdir/inter_30_hists.m -q 30 $outputdir/merged_nodups.txt $outputdir/inter_30.hic $genomePath
