hifiasm是一个能有效利用PacBio HiFi测序技术,在分型组装图(pahsed assembly gprah)中可靠的表示单倍体信息的算法。
流程介绍
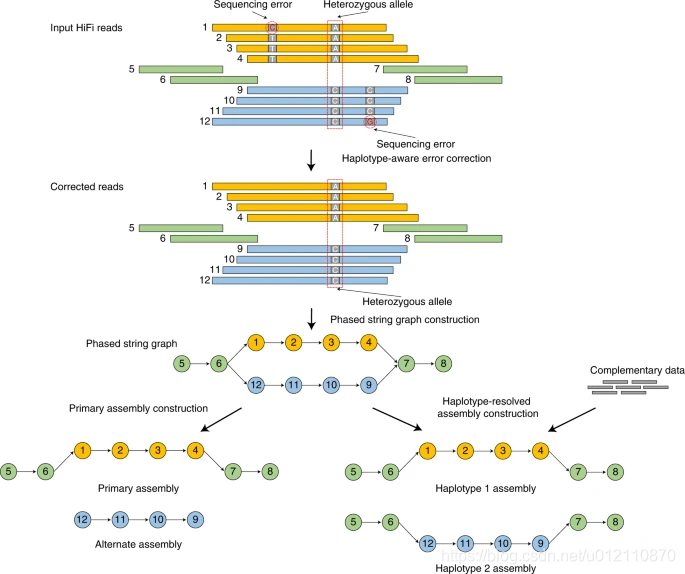
hifiasm的分析流程如下,主要分为3个阶段
第一阶段:通过所有序列的相互比对,对前在测序错误进行纠正。如果一个位置只存在两种碱基类型,且每个碱基类型至少有3条read支持,那么这个位置会被当作杂合位点,否则,视作测序错误,将被纠正。
第二阶段:根据序列之间的重叠关系,构建分型的字符串图(phased string graph)。其中调整朝向的序列作为顶点(vertex),一致重叠作为边(edge)。字符串图中的气泡(bubble)则是杂合位点。
第三阶段:如果没有额外的信息,hifiasm会随机选择气泡的一边构建primary assembly,另一边则是alternate assembly. 该策略和HiCanu,Falcon-Unzip一样。对于杂合基因组而言,由于存在一个以上的纯合haplotype,因此primary assembly可能还会包含haplotigs。HiCanu依赖于第三方的purge_dups, 而hifiasm内部实现了purge_dups算法的变种,简化了流程。如果有额外的信息,那么hifiasm就可以正确的对haplotype进行分型。
安装
hifiasm仅仅依赖 g++和zlib,以及git
# 依赖g++和zlib
git clone https://github.com/chhylp123/hifiasm
cd hifiasm && make
# bioconda
conda install -c bioconda hifiasm
通过源码编译的方式安装,需要将hifiasm移动到你的软件目录下,或者将hifiasm的路径加入到环境变量PATH中。
如果是 trio-binning模式,需要额外安装yak
#source code
git clone https://github.com/lh3/yak
cd yak && make
# bioncda
conda install -c bioconda yak
案例展示
hifiasm的使用非常简洁明了,根据已有的数据分为,仅HiFi数据模式,有双亲二代测序的Trio-binning模式和有Hi-C数据的Hi-C模式。
仅有HiFi数据
最基本的用法,会得到两个部分分型的组装
wget https://github.com/chhylp123/hifiasm/releases/download/v0.7/chr11-2M.fa.gz
hifiasm -o test -t 32 chr11-2M.fa.gz 2> test.log
其中 -o定义输出文件的文件名前缀, -t是线程数
运行结束后生成的一系列文件中,我们只需要关注如下几项 (prefix表示前缀)
-
prefix.bp.r_utg.gfa: haplotype-resolved raw unitig graph,记录所有的单倍型信息 -
prefix.bp.p_utg.gfa: 在raw unitig graph基础上过滤小的bubble, -
prefix.bp.p_ctg.gfa: 主要contig的assembly graph -
prefix.bp.hap1.p_ctg.gfa: haplotype1的部分分型的contig graph -
prefix.bp.hap2.p_ctg.gfa: haplotype2的部分分型的contig graph
如果并不需要部分分型的组装,而只想要primary和alternate的组装结果,可以在之前的命令的基础上,加上 --primary参数。
hifiasm --primary -o test -t 32 chr11-2M.fa.gz 2> test.log2
由于hifiasm运行时会将步骤中纠错和相互比对的结果保存成 bin 文件,因此重新这一次运行速度会很快
primay模式下输出的文件和之前的类似,唯一的不同在于没有 bp
-
``prefix
.r_utg.gfa: haplotype-resolved raw unitig graph -
``prefix
.p_utg.gfa: haplotype-resolved processed unitig graph without small bubbles. -
``prefix
.p_ctg.gfa: assembly graph of primary contigs. -
``prefix
.a_ctg.gfa: assembly graph of alternate contigs.
我们关心的,可能就是 主要的contig,通过awk进行提取
awk '/^S/{print ">"$2;print $3}' test.p_ctg.gfa > test.p_ctg.fa
Trio-binning模式
如果测了双亲,则可以使用trio-binning方法进行更加可靠的分型。分为两步,先用yak统计k-mers, 然后用hifiasm进行组装
案例代码如下
yak count -k31 -b37 -t16 -o pat.yak paternal.fq.gz
yak count -k31 -b37 -t16 -o mat.yak maternal.fq.gz
hifiasm -o NA12878.asm -t 32 -1 pat.yak -2 mat.yak NA12878.fq.gz
输出的文章和之前类似,主要关心其中文件名带 dip 的输出gfa文件
-
prefix.dip.hap1.p_ctg.gfa: 完成分型的父源单倍体 contig图. -
prefix.dip.hap2.p_ctg.gfa: 完全分型的母源单倍体contig图.
整合Hi-C数据
由于Hi-C数据能够提供远距信息,因此也能用于单倍体分型。只需要加上两个参数, h1接受Hi-C的read1, h2 接受Hi-C的read2
hifiasm -o NA12878.asm -t32 --h1 read1.fq.gz --h2 read2.fq.gz HiFi-reads.fq.gz
在该模式下,每个contig要么是来自于父亲,要么是来自于母亲。hifiasm会将同一来源的contig放在同一个组装中。需要注意的是,hifiasm未必能够处理好着丝粒附近的区域,另外hifiasm中Hi-C也不会用于进行scaffold。
输出结果中,我们重点关注其中名字带hic的文件
-
prefix.hic.p_ctg.gfa: 主要contig的组装图 -
prefix.hic.hap1.p_ctg.gfa: 完全分型的haplotype1的contig图 -
prefix.hic.hap2.p_ctg.gfa: 完全分型的haplotype2的contig图 -
prefix.hic.a_ctg.gfa: 如果设置了--primary参数,还会输出该次要contig的组装图
日志和参数调整
绝大部分的时候,我们只需要使用默认参数即可得到相对比较好的结果。但是当默认参数无法达到自己的目的,那我们就需要检查日志信息,阅读相关参数从而优化结果。
日志信息主要分为三项
-
k-mer图: 纯合样本只有一个peak,杂合样本则会有2个peak。
-
纯合峰的覆盖度:
[M::purge_dups] homozygous read coverage threshold: X, 一般会由hifiasm自动推断。 -
杂合/纯合碱基数目(Hi-C模式): 在Hi-C模式下,如果纯合的碱基数超过杂合碱基数,那么hifiasm就不容易找对纯合read的所在峰。
对于日志信息,我们最主要关注的就是k-mer图,从而判断hifiasm是否能够正确的找到纯合峰,杂合峰的所在位置。如果hifiasm没有找对纯合峰所在的位置,就需要我们根据k-mer图手动指定 --hom-cov。
对于一个组装结果,最直接的评估标准就是基因组大小是否符合预期,分型的两套基因组是否相差不大,序列是否足够长,是否存在错误组装的情况。
如果基因组大小不符合预期,一般都是hifiasm找错了纯合峰的位置,我们需要手动指定 --hom-cov;如果分型的两套基因组差别过大,则通过降低 -s 调整。如果序列不够长,片段化明显,则可以尝试增加 -D 和 -N, 虽然会增加运行时间,但是会提高重复区域的分辨率。如果后续的Hi-C,或者BioNano发现hifiasm组装结果有比较多错误组装,则可以适当降低 --purge-max, -s和 -O。或者设置 -u 关闭post-join 步骤,hifiasm通过该步骤提高组装的连续性。
参考资料
https://github.com/chhylp123/hifiasm
