chromVAR: 从单细胞表观数据中推断转录因子相关开放区域
chromVAR是一个用于分析稀疏染色质开放的R包。chromVAR的输入文件包括,ATAC-seq处理后的fragments文件(过滤重复和低质量数据), DNAse-seq实验结果,以及基因组注释(例如motif位置)
从零开始学CIRCOS绘制圈图(五)
这一部分承接从零开始学CIRCOS绘制圈图(四),对之前的布局进行调整,使结构更加适合于发表。染色体的位置顺序默认情况下是会显示所有的染色体,而且会先从ath1到ath5,然后从aly1到aly8, 那么如何调整顺序呢?需要设置的参数是chromosomes_order, 按照自己的需求调整位置。
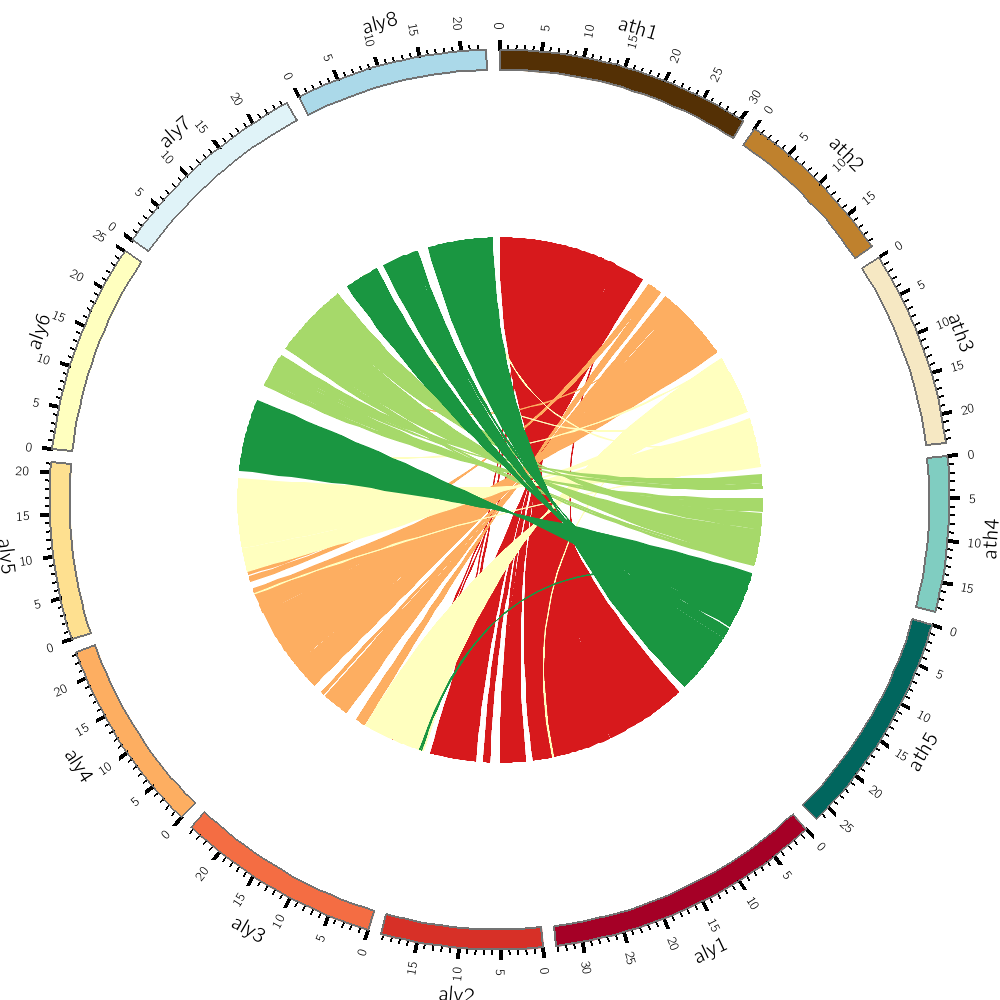
从零开始学CIRCOS绘制圈图(四)
通常circos的中间部分不是空白区域,会用一条条线进行连接,表示两个染色体部分区域有关系。数据格式对于link,circos要求输入数据至少有6列,分别是chr1 start1 end1 chr2 start2 end2 [options]举个例子chr110000002000000chr5300